npj Computational Materials ( IF 9.4 ) Pub Date : 2024-09-12 , DOI: 10.1038/s41524-024-01374-8 Logan Ward , Ben Blaiszik , Cheng-Wei Lee , Troy Martin , Ian Foster , André Schleife
|
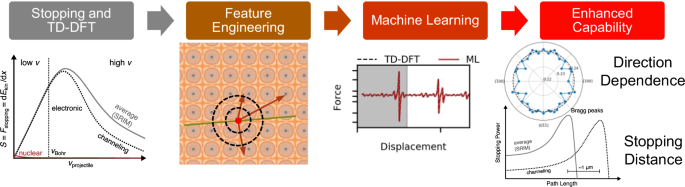
Knowing the rate at which particle radiation releases energy in a material, the “stopping power,” is key to designing nuclear reactors, medical treatments, semiconductor and quantum materials, and many other technologies. While the nuclear contribution to stopping power, i.e., elastic scattering between atoms, is well understood in the literature, the route for gathering data on the electronic contribution has for decades remained costly and reliant on many simplifying assumptions, including that materials are isotropic. We establish a method that combines time-dependent density functional theory (TDDFT) and machine learning to reduce the time to assess new materials to hours on a supercomputer and provide valuable data on how atomic details influence electronic stopping. Our approach uses TDDFT to compute the electronic stopping from first principles in several directions and then machine learning to interpolate to other directions at a cost of 10 million times fewer core-hours. We demonstrate the combined approach in a study of proton irradiation in aluminum and employ it to predict how the depth of maximum energy deposition, the “Bragg Peak,” varies depending on the incident angle—a quantity otherwise inaccessible to modelers and far outside the scales of quantum mechanical simulations. The lack of any experimental information requirement makes our method applicable to most materials, and its speed makes it a prime candidate for enabling quantum-to-continuum models of radiation damage. The prospect of reusing valuable TDDFT data for training the model makes our approach appealing for applications in the age of materials data science.
中文翻译:
利用瞬态密度泛函理论和机器学习加速多尺度电子制动力预测
了解粒子辐射在材料中释放能量的速率(即“阻止能力”)是设计核反应堆、医疗、半导体和量子材料以及许多其他技术的关键。虽然核对阻止能力的贡献,即原子之间的弹性散射,在文献中得到了很好的理解,但几十年来收集电子贡献数据的途径仍然成本高昂,并且依赖于许多简化的假设,包括材料是各向同性的。我们建立了一种结合时间相关密度泛函理论(TDDFT)和机器学习的方法,将超级计算机上评估新材料的时间减少到几个小时,并提供有关原子细节如何影响电子停止的有价值的数据。我们的方法使用 TDDFT 从多个方向的第一原理计算电子停止,然后通过机器学习插值到其他方向,成本减少了 1000 万倍的核心时间。我们在铝质子辐照研究中展示了这种组合方法,并用它来预测最大能量沉积的深度(“布拉格峰”)如何随入射角变化——否则建模者无法获得该量,而且远远超出了尺度的量子力学模拟。由于缺乏任何实验信息要求,我们的方法适用于大多数材料,并且其速度使其成为实现辐射损伤的量子到连续模型的主要候选者。重用有价值的 TDDFT 数据来训练模型的前景使我们的方法在材料数据科学时代的应用中具有吸引力。






























 京公网安备 11010802027423号
京公网安备 11010802027423号