当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Effect of Protein-Polarized Ligand Charges on Relative Protein Ligand Binding Affinities
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-11 , DOI: 10.1021/acs.jctc.3c01337 Suliman Adam 1 , Itamar Kass 1 , Dana Krepel-Zussman 1 , Gal Masarati 1 , Dorit Shemesh 1 , Avital Sharir-Ivry 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-11 , DOI: 10.1021/acs.jctc.3c01337 Suliman Adam 1 , Itamar Kass 1 , Dana Krepel-Zussman 1 , Gal Masarati 1 , Dorit Shemesh 1 , Avital Sharir-Ivry 1
Affiliation
|
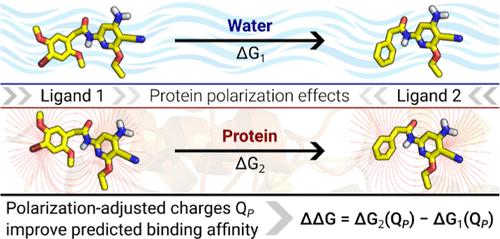
A major challenge in computer-aided drug design is predicting relative binding energies of different molecules to a target protein using fast and accurate free-energy calculation methods. Free-energy calculations are primarily computed by utilizing classical molecular dynamics simulations based on all-atom force fields (FF) to model the interactions in the system. The present standard classical all-atom FFs contain fixed partial charges on the atoms, and hence electrostatic interactions are modeled between them. The parametrization process to determine these partial charges usually relies on quantum mechanics or semiempirical calculations of the molecule in the gas phase or homogeneous water surrounding. These present standard parametrization schemes of the partial charges neglect, therefore, polarization effects from the protein surrounding. The absence of protein polarization effects can lead to significant errors in free-energy calculations in proteins. We present a parametrization scheme for the partial charges of ligands, named protein-induced polarization (PIP) charges, which account for the electrostatic polarization due to the protein surrounding. The scheme involves single-point quantum mechanics/molecular mechanics calculations of the ligand charges in the protein/water surrounding. Using PIP ligand partial charges, we have calculated the relative binding free energies (RBFEs) of well-studied protein−ligand systems. We show here that RBFEs computed with PIP charges are either significantly improved or at least comparable to those computed with nonpolarized standard GAFF charges. Overall, we present a simple-to-use parametrization scheme to include protein polarization in any type of binding free-energy calculations. The parametrization scheme increases the accuracy in RBFE calculations, while it does not add significant computation time to standard parametrization procedures.
中文翻译:

蛋白质极化配体电荷对相关蛋白质配体结合亲和力的影响
计算机辅助药物设计的一个主要挑战是使用快速准确的自由能计算方法预测不同分子与靶蛋白的相对结合能。自由能计算主要通过利用基于全原子力场 (FF) 的经典分子动力学模拟来模拟系统中的相互作用。目前的标准经典全原子 FF 在原子上包含固定的部分电荷,因此对它们之间的静电相互作用进行了建模。确定这些部分电荷的参数化过程通常依赖于气相或均匀水周围的分子的量子力学或半经验计算。因此,这些目前的部分电荷的标准参数化方案忽略了来自蛋白质周围的极化效应。蛋白质极化效应的缺失可能会导致蛋白质自由能计算出现重大错误。我们提出了配体部分电荷的参数化方案,称为蛋白质诱导极化(PIP)电荷,它解释了蛋白质周围引起的静电极化。该方案涉及蛋白质/水周围配体电荷的单点量子力学/分子力学计算。使用 PIP 配体部分电荷,我们计算了经过充分研究的蛋白质-配体系统的相对结合自由能 (RBFE)。我们在这里表明,使用 PIP 电荷计算的 RBFE 要么显着改善,要么至少与使用非极化标准 GAFF 电荷计算的 RBFE 相当。总的来说,我们提出了一种简单易用的参数化方案,将蛋白质极化纳入任何类型的结合自由能计算中。 参数化方案提高了 RBFE 计算的准确性,同时不会给标准参数化过程增加大量计算时间。
更新日期:2024-09-11
中文翻译:
蛋白质极化配体电荷对相关蛋白质配体结合亲和力的影响
计算机辅助药物设计的一个主要挑战是使用快速准确的自由能计算方法预测不同分子与靶蛋白的相对结合能。自由能计算主要通过利用基于全原子力场 (FF) 的经典分子动力学模拟来模拟系统中的相互作用。目前的标准经典全原子 FF 在原子上包含固定的部分电荷,因此对它们之间的静电相互作用进行了建模。确定这些部分电荷的参数化过程通常依赖于气相或均匀水周围的分子的量子力学或半经验计算。因此,这些目前的部分电荷的标准参数化方案忽略了来自蛋白质周围的极化效应。蛋白质极化效应的缺失可能会导致蛋白质自由能计算出现重大错误。我们提出了配体部分电荷的参数化方案,称为蛋白质诱导极化(PIP)电荷,它解释了蛋白质周围引起的静电极化。该方案涉及蛋白质/水周围配体电荷的单点量子力学/分子力学计算。使用 PIP 配体部分电荷,我们计算了经过充分研究的蛋白质-配体系统的相对结合自由能 (RBFE)。我们在这里表明,使用 PIP 电荷计算的 RBFE 要么显着改善,要么至少与使用非极化标准 GAFF 电荷计算的 RBFE 相当。总的来说,我们提出了一种简单易用的参数化方案,将蛋白质极化纳入任何类型的结合自由能计算中。 参数化方案提高了 RBFE 计算的准确性,同时不会给标准参数化过程增加大量计算时间。






























 京公网安备 11010802027423号
京公网安备 11010802027423号