当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hybrid Quantum Mechanical, Molecular Mechanical, and Machine Learning Potential for Computing Aqueous-Phase Adsorption Free Energies on Metal Surfaces
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-10 , DOI: 10.1021/acs.jctc.4c00869 Mehdi Zare 1 , Dia Sahsah 1 , Mohammad Saleheen 1 , Jörg Behler 2, 3 , Andreas Heyden 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-10 , DOI: 10.1021/acs.jctc.4c00869 Mehdi Zare 1 , Dia Sahsah 1 , Mohammad Saleheen 1 , Jörg Behler 2, 3 , Andreas Heyden 1
Affiliation
|
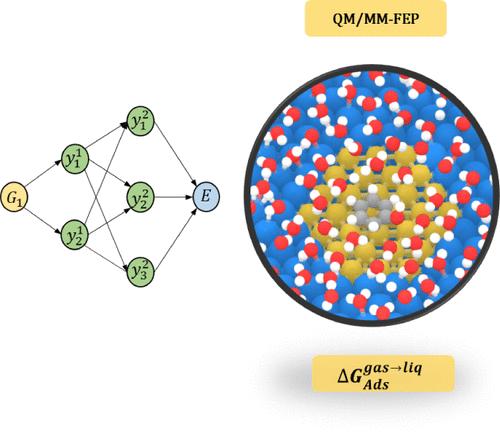
Performing reliable computer simulations of elementary processes occurring at metal–water interfaces is pivotal for novel catalyst design in sustainable energy applications. Computational catalyst design hinges on the ability to reliably and efficiently compute the potential energy surface (PES) of the system. Due to the large system sizes needed for studying processes at liquid water–metal interfaces, these systems can currently not be described using density functional theory (DFT). In this work, we used a hybrid quantum mechanical, molecular mechanical, and machine learning potential for studying the adsorption behavior of phenol, atomic hydrogen, 2-butanol, and 2-butanone on the (0001) facet of Ru under reducing conditions when Ru is not oxidized. Specifically, we describe the adsorbate and the surrounding metal atoms at the DFT level of theory. Here, we also considered the electrostatic field effect of the water molecules on adsorbate–metal interactions. Next, for the water–water and water–adsorbate interactions, we used established classical force fields. Finally, for the water–Ru surface interaction, for which no reliable force fields have been published, we used Behler–Parrinello high-dimensional neural network potentials (HDNNPs). Employing this setup, we used our explicit solvation for metal surface (eSMS) approach to compute the aqueous-phase effect on the low-coverage adsorption of selected molecules and atoms on the (0001) facet of Ru. In agreement with previous experimental and computational studies of oxygenated molecules over transition metal facets, we found that liquid water destabilizes the tested adsorbates on Ru(0001). Interestingly, our findings indicate that adsorbates on Ru are less affected by the presence of an aqueous phase than on other transition metals (e.g., Pt), highlighting the necessity of experimental investigations of Ru-based catalytic systems in liquid water.
中文翻译:

计算金属表面水相吸附自由能的混合量子力学、分子力学和机器学习潜力
对金属-水界面发生的基本过程进行可靠的计算机模拟对于可持续能源应用中的新型催化剂设计至关重要。计算催化剂设计取决于可靠且高效地计算系统势能面 (PES) 的能力。由于研究液态水-金属界面的过程需要较大的系统尺寸,目前无法使用密度泛函理论(DFT)来描述这些系统。在这项工作中,我们使用混合量子力学、分子力学和机器学习势来研究当 Ru 还原条件下苯酚、原子氢、2-丁醇和 2-丁酮在 Ru 的 (0001) 面上的吸附行为。没有被氧化。具体来说,我们在 DFT 理论水平上描述了吸附物和周围的金属原子。在这里,我们还考虑了水分子的静电场效应对吸附物-金属相互作用的影响。接下来,对于水-水和水-吸附物相互作用,我们使用已建立的经典力场。最后,对于水-钌表面相互作用(尚未发表可靠的力场),我们使用了 Behler-Parrinello 高维神经网络势(HDNNP)。采用这种设置,我们使用金属表面显式溶剂化 (eSMS) 方法来计算水相对 Ru (0001) 面上选定分子和原子的低覆盖度吸附的影响。与之前对过渡金属面上含氧分子的实验和计算研究一致,我们发现液态水使 Ru(0001) 上测试的吸附物不稳定。 有趣的是,我们的研究结果表明,与其他过渡金属(例如 Pt)相比,Ru 上的吸附物受水相存在的影响较小,这凸显了对液态水中的 Ru 基催化系统进行实验研究的必要性。
更新日期:2024-09-10
中文翻译:
计算金属表面水相吸附自由能的混合量子力学、分子力学和机器学习潜力
对金属-水界面发生的基本过程进行可靠的计算机模拟对于可持续能源应用中的新型催化剂设计至关重要。计算催化剂设计取决于可靠且高效地计算系统势能面 (PES) 的能力。由于研究液态水-金属界面的过程需要较大的系统尺寸,目前无法使用密度泛函理论(DFT)来描述这些系统。在这项工作中,我们使用混合量子力学、分子力学和机器学习势来研究当 Ru 还原条件下苯酚、原子氢、2-丁醇和 2-丁酮在 Ru 的 (0001) 面上的吸附行为。没有被氧化。具体来说,我们在 DFT 理论水平上描述了吸附物和周围的金属原子。在这里,我们还考虑了水分子的静电场效应对吸附物-金属相互作用的影响。接下来,对于水-水和水-吸附物相互作用,我们使用已建立的经典力场。最后,对于水-钌表面相互作用(尚未发表可靠的力场),我们使用了 Behler-Parrinello 高维神经网络势(HDNNP)。采用这种设置,我们使用金属表面显式溶剂化 (eSMS) 方法来计算水相对 Ru (0001) 面上选定分子和原子的低覆盖度吸附的影响。与之前对过渡金属面上含氧分子的实验和计算研究一致,我们发现液态水使 Ru(0001) 上测试的吸附物不稳定。 有趣的是,我们的研究结果表明,与其他过渡金属(例如 Pt)相比,Ru 上的吸附物受水相存在的影响较小,这凸显了对液态水中的 Ru 基催化系统进行实验研究的必要性。
































 京公网安备 11010802027423号
京公网安备 11010802027423号