当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Developing and Benchmarking Sulfate and Sulfamate Force Field Parameters via Ab Initio Molecular Dynamics Simulations To Accurately Model Glycosaminoglycan Electrostatic Interactions
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-09-09 , DOI: 10.1021/acs.jcim.4c00981 Miguel Riopedre-Fernandez 1 , Vojtech Kostal 1 , Tomas Martinek 1 , Hector Martinez-Seara 1 , Denys Biriukov 1, 2, 3
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-09-09 , DOI: 10.1021/acs.jcim.4c00981 Miguel Riopedre-Fernandez 1 , Vojtech Kostal 1 , Tomas Martinek 1 , Hector Martinez-Seara 1 , Denys Biriukov 1, 2, 3
Affiliation
|
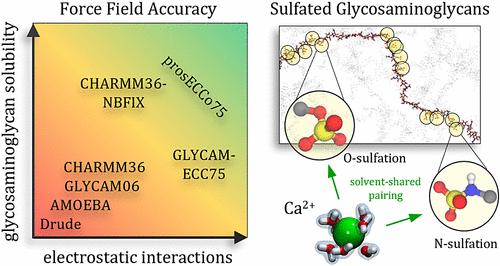
Glycosaminoglycans (GAGs) are negatively charged polysaccharides found on cell surfaces, where they regulate transport pathways of foreign molecules toward the cell. The structural and functional diversity of GAGs is largely attributed to varied sulfation patterns along the polymer chains, which makes understanding their molecular recognition mechanisms crucial. Molecular dynamics (MD) simulations, thanks to their unmatched microscopic resolution, have the potential to be a reference tool for exploring the patterns responsible for biologically relevant interactions. However, the capability of molecular dynamics force fields used in biosimulations to accurately capture sulfation-specific interactions is not well established, partly due to the intrinsic properties of GAGs that pose challenges for most experimental techniques. In this work, we evaluate the performance of molecular dynamics force fields for sulfated GAGs by studying ion pairing of Ca2+ to sulfated moieties─N-methylsulfamate and methylsulfate─that resemble N- and O-sulfation found in GAGs, respectively. We tested available nonpolarizable (CHARMM36 and GLYCAM06) and explicitly polarizable (Drude and AMOEBA) force fields, and derived new implicitly polarizable models through charge scaling (prosECCo75 and GLYCAM-ECC75) that are consistent with our developed “charge-scaling” framework. The calcium–sulfamate/sulfate interaction free energy profiles obtained with the tested force fields were compared against reference ab initio molecular dynamics (AIMD) simulations, which serve as a robust alternative to experiments. AIMD simulations indicate that the preferential Ca2+ binding mode to sulfated GAG groups is solvent-shared pairing. Only our scaled-charge models agree satisfactorily with the AIMD data, while all other force fields exhibit poorer agreement, sometimes even qualitatively. Surprisingly, even explicitly polarizable force fields display a notable disagreement with the AIMD data, likely attributed to difficulties in their optimization and possible inherent limitations in depicting high-charge-density ion interactions accurately. Finally, the underperforming force fields lead to unrealistic aggregation of sulfated saccharides, which qualitatively disagrees with our understanding of the soft glycocalyx environment. Our results highlight the importance of accurately treating electronic polarization in MD simulations of sulfated GAGs and caution against over-reliance on currently available models without thorough validation and optimization.
中文翻译:

通过从头算分子动力学模拟开发硫酸盐和氨基磺酸盐力场参数并对其进行基准测试,以准确模拟糖胺聚糖静电相互作用
糖胺聚糖 (GAG) 是在细胞表面发现的带负电荷的多糖,它们调节外来分子向细胞的运输途径。 GAG 的结构和功能多样性很大程度上归因于聚合物链上不同的硫酸化模式,这使得了解其分子识别机制至关重要。分子动力学(MD)模拟由于其无与伦比的微观分辨率,有可能成为探索生物相关相互作用模式的参考工具。然而,生物模拟中使用的分子动力学力场准确捕获硫酸化特异性相互作用的能力尚未得到充分证实,部分原因是 GAG 的固有特性给大多数实验技术带来了挑战。在这项工作中,我们通过研究 Ca 2+与硫酸化部分( N-甲基氨基磺酸盐和甲基硫酸盐)的离子配对来评估硫酸化 GAG 的分子动力学力场性能,这些硫酸化部分分别类似于 GAG 中的 N- 和 O-硫酸化。我们测试了可用的不可极化(CHARMM36 和 GLYCAM06)和显式极化(Drude 和 AMOEBA)力场,并通过电荷缩放(prosECCo75 和 GLYCAM-ECC75)导出了新的隐式极化模型,这些模型与我们开发的“电荷缩放”框架一致。将通过测试力场获得的钙-氨基磺酸盐/硫酸盐相互作用自由能分布与参考从头分子动力学 (AIMD) 模拟进行比较,该模拟可作为实验的可靠替代方案。 AIMD 模拟表明,Ca 2+与硫酸化GAG 基团的优先结合模式是溶剂共享配对。 只有我们的缩放电荷模型与 AIMD 数据令人满意地一致,而所有其他力场的一致性较差,有时甚至是定性的。令人惊讶的是,即使是明确的极化力场也显示出与 AIMD 数据的显着不一致,这可能归因于其优化的困难以及准确描述高电荷密度离子相互作用的可能固有限制。最后,表现不佳的力场导致硫酸化糖的不切实际的聚集,这在质量上与我们对软糖萼环境的理解不符。我们的结果强调了在硫酸化 GAG 的 MD 模拟中准确处理电子极化的重要性,并警告不要过度依赖当前可用的模型而不进行彻底的验证和优化。
更新日期:2024-09-09
中文翻译:
通过从头算分子动力学模拟开发硫酸盐和氨基磺酸盐力场参数并对其进行基准测试,以准确模拟糖胺聚糖静电相互作用
糖胺聚糖 (GAG) 是在细胞表面发现的带负电荷的多糖,它们调节外来分子向细胞的运输途径。 GAG 的结构和功能多样性很大程度上归因于聚合物链上不同的硫酸化模式,这使得了解其分子识别机制至关重要。分子动力学(MD)模拟由于其无与伦比的微观分辨率,有可能成为探索生物相关相互作用模式的参考工具。然而,生物模拟中使用的分子动力学力场准确捕获硫酸化特异性相互作用的能力尚未得到充分证实,部分原因是 GAG 的固有特性给大多数实验技术带来了挑战。在这项工作中,我们通过研究 Ca 2+与硫酸化部分( N-甲基氨基磺酸盐和甲基硫酸盐)的离子配对来评估硫酸化 GAG 的分子动力学力场性能,这些硫酸化部分分别类似于 GAG 中的 N- 和 O-硫酸化。我们测试了可用的不可极化(CHARMM36 和 GLYCAM06)和显式极化(Drude 和 AMOEBA)力场,并通过电荷缩放(prosECCo75 和 GLYCAM-ECC75)导出了新的隐式极化模型,这些模型与我们开发的“电荷缩放”框架一致。将通过测试力场获得的钙-氨基磺酸盐/硫酸盐相互作用自由能分布与参考从头分子动力学 (AIMD) 模拟进行比较,该模拟可作为实验的可靠替代方案。 AIMD 模拟表明,Ca 2+与硫酸化GAG 基团的优先结合模式是溶剂共享配对。 只有我们的缩放电荷模型与 AIMD 数据令人满意地一致,而所有其他力场的一致性较差,有时甚至是定性的。令人惊讶的是,即使是明确的极化力场也显示出与 AIMD 数据的显着不一致,这可能归因于其优化的困难以及准确描述高电荷密度离子相互作用的可能固有限制。最后,表现不佳的力场导致硫酸化糖的不切实际的聚集,这在质量上与我们对软糖萼环境的理解不符。我们的结果强调了在硫酸化 GAG 的 MD 模拟中准确处理电子极化的重要性,并警告不要过度依赖当前可用的模型而不进行彻底的验证和优化。






























 京公网安备 11010802027423号
京公网安备 11010802027423号