当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical kinetic studies on intramolecular H-migration reactions of peroxy radicals of diethoxymethane
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-09-10 , DOI: 10.1039/d4cp02302a Siyu Chen , Juan-Qin Li , Quan Zhu , Zerong Li
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-09-10 , DOI: 10.1039/d4cp02302a Siyu Chen , Juan-Qin Li , Quan Zhu , Zerong Li
|
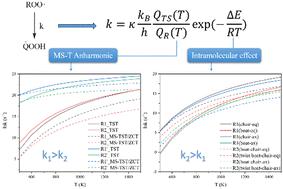
Diethoxymethane (DEM), a promising carbon-neutral fuel, has high reactivity at low temperatures. The intramolecular hydrogen migration reaction of the DEM peroxy radicals can be viewed as a critical step in the low temperature oxidation mechanism of DEM. In this work, multistructural transition state theory (MS-TST) was utilized to calculate the high-pressure limit rate constants of 1,5, 1,6 and 1,7 H-migration reactions for DEM peroxy radicals. In addition to the tunneling effects and anharmonic effects, the intramolecular effects, including steric hindrance, intramolecular hydrogen bonding and conformational changes in reactants and transition states, are also considered in the rate constant calculations. The calculated energy barriers and rate constants demonstrated the substantial impact of intramolecular effects on the kinetics of H-migration reactions in DEM peroxy radicals. Specifically, the distinct configurations of transition states could potentially influence the reaction kinetics. The pressure-dependent rate constants are computed using system-specific quantum RRK theory. The calculated results show that the falloff effect of 1,5 and 1,6 H-migration reactions is more pronounced than that of the 1,7 H-migration reaction. The thermodynamics and kinetics presented in this study could be instrumental in understanding the low-temperature oxidation mechanism of DEM and might prove crucial for future research on comprehensively analyzing the autoignition behavior.
中文翻译:

二乙氧基甲烷过氧自由基分子内氢迁移反应的理论动力学研究
二乙氧基甲烷(DEM)是一种很有前景的碳中性燃料,在低温下具有高反应性。 DEM过氧自由基的分子内氢迁移反应可以被视为DEM低温氧化机理的关键步骤。在这项工作中,利用多结构过渡态理论(MS-TST)计算了DEM过氧自由基的1,5、1,6和1,7 H-迁移反应的高压极限速率常数。除了隧道效应和非简谐效应外,在速率常数计算中还考虑了分子内效应,包括空间位阻、分子内氢键以及反应物和过渡态的构象变化。计算的能垒和速率常数证明了分子内效应对 DEM 过氧自由基中 H 迁移反应动力学的重大影响。具体来说,过渡态的不同构型可能会影响反应动力学。压力相关的速率常数是使用系统特定的量子 RRK 理论计算的。计算结果表明,1,5和1,6 H-迁移反应的衰减效应比1,7 H-迁移反应更明显。本研究中提出的热力学和动力学有助于理解 DEM 的低温氧化机理,并且可能对未来全面分析自燃行为的研究至关重要。
更新日期:2024-09-10
中文翻译:
二乙氧基甲烷过氧自由基分子内氢迁移反应的理论动力学研究
二乙氧基甲烷(DEM)是一种很有前景的碳中性燃料,在低温下具有高反应性。 DEM过氧自由基的分子内氢迁移反应可以被视为DEM低温氧化机理的关键步骤。在这项工作中,利用多结构过渡态理论(MS-TST)计算了DEM过氧自由基的1,5、1,6和1,7 H-迁移反应的高压极限速率常数。除了隧道效应和非简谐效应外,在速率常数计算中还考虑了分子内效应,包括空间位阻、分子内氢键以及反应物和过渡态的构象变化。计算的能垒和速率常数证明了分子内效应对 DEM 过氧自由基中 H 迁移反应动力学的重大影响。具体来说,过渡态的不同构型可能会影响反应动力学。压力相关的速率常数是使用系统特定的量子 RRK 理论计算的。计算结果表明,1,5和1,6 H-迁移反应的衰减效应比1,7 H-迁移反应更明显。本研究中提出的热力学和动力学有助于理解 DEM 的低温氧化机理,并且可能对未来全面分析自燃行为的研究至关重要。






























 京公网安备 11010802027423号
京公网安备 11010802027423号