当前位置:
X-MOL 学术
›
Adv. Energy Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Controllable In situ Polymerization of 1,3-Dioxolane via Sustained-Release Effect for Solid-State Lithium Metal Batteries
Advanced Energy Materials ( IF 24.4 ) Pub Date : 2024-09-09 , DOI: 10.1002/aenm.202402848 Sucheng Liu 1 , Boyong Wu 1 , Song Huang 2 , Zitian Lin 1 , Huiyu Song 1 , Li Du 1 , Zhenxing Liang 1 , Zhiming Cui 1
Advanced Energy Materials ( IF 24.4 ) Pub Date : 2024-09-09 , DOI: 10.1002/aenm.202402848 Sucheng Liu 1 , Boyong Wu 1 , Song Huang 2 , Zitian Lin 1 , Huiyu Song 1 , Li Du 1 , Zhenxing Liang 1 , Zhiming Cui 1
Affiliation
|
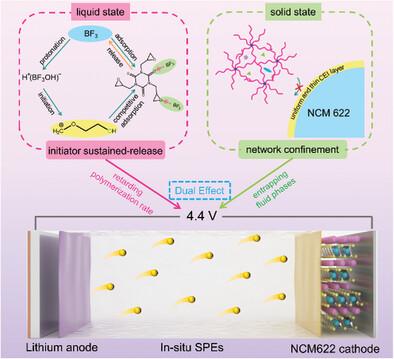
In situ formed poly(1,3-dioxolane) (PDOL) electrolytes are of great interest due to the facile process and the improved interface contact. However, the practical application of in situ PDOL electrolytes is still plagued by fast solidification time (liquid state) and poor high-voltage stability (solid state). Herein, the slow-release carriers triglycidyl isocyanurate (TGIC), which play dual roles as initiator sustained-release and network confinement, can tune DOL curing time and cathode/electrolyte interface chemistry is demonstrated. Specifically, the electronegative C≐O and epoxy groups in TGIC have an affinity with BF3, the decomposition product of lithium bis(oxalate)borate (LiDFOB), delaying BF3 protonation reaction and thus extending DOL solidification time. In addition, the epoxy groups in TGIC serve as crosslinking sites to form in situ crosslinked polymer electrolytes (TPDOL@FEC). The corresponding network structure suppresses the contact reaction between high-fluidity organic components and cathodes, generating a uniform and thin cathode electrolyte interface layer. As a result, the TPDOL@FEC precursor solution can remain its liquid state even after resting 24 h at room temperature. The assembled LiNi0.6Co0.2Mn0.2O2||TPDOL@FEC||Li cells display an impressive capacity retention of 91.5% after 100 cycles at 4.4 V (0.5 C). This study is expected to be a leap in the pursuit of practically feasible in situ formed PDOL electrolytes.
中文翻译:

固态锂金属电池中1,3-二氧戊环缓释效应的可控原位聚合
原位形成的聚(1,3-二氧戊环)(PDOL)电解质由于其简便的工艺和改进的界面接触而引起了人们的极大兴趣。然而,原位PDOL电解质的实际应用仍然受到凝固时间快(液态)和高压稳定性差(固态)的困扰。在此,缓释载体异氰脲酸三缩水甘油酯(TGIC)具有引发剂缓释和网络限制的双重作用,可以调节DOL固化时间,并证明了阴极/电解质界面化学。具体而言,TGIC中的负电性C≐O和环氧基团与双(草酸)硼酸锂(LiDFOB)的分解产物BF 3具有亲和力,延迟了BF 3质子化反应,从而延长了DOL固化时间。此外,TGIC中的环氧基团作为交联位点形成原位交联聚合物电解质(TPDOL@FEC)。相应的网络结构抑制了高流动性有机组分与正极之间的接触反应,产生均匀且薄的正极电解质界面层。因此,即使在室温下静置 24 小时后,TPDOL@FEC 前体溶液仍能保持液态。组装好的 LiNi 0.6 Co 0.2 Mn 0.2 O 2 ||TPDOL@FEC||Li 电池在 4.4 V (0.5 C) 下循环 100 次后,容量保持率高达 91.5%。这项研究预计将成为追求实际可行的原位形成的 PDOL 电解质的一个飞跃。
更新日期:2024-09-09
中文翻译:
固态锂金属电池中1,3-二氧戊环缓释效应的可控原位聚合
原位形成的聚(1,3-二氧戊环)(PDOL)电解质由于其简便的工艺和改进的界面接触而引起了人们的极大兴趣。然而,原位PDOL电解质的实际应用仍然受到凝固时间快(液态)和高压稳定性差(固态)的困扰。在此,缓释载体异氰脲酸三缩水甘油酯(TGIC)具有引发剂缓释和网络限制的双重作用,可以调节DOL固化时间,并证明了阴极/电解质界面化学。具体而言,TGIC中的负电性C≐O和环氧基团与双(草酸)硼酸锂(LiDFOB)的分解产物BF 3具有亲和力,延迟了BF 3质子化反应,从而延长了DOL固化时间。此外,TGIC中的环氧基团作为交联位点形成原位交联聚合物电解质(TPDOL@FEC)。相应的网络结构抑制了高流动性有机组分与正极之间的接触反应,产生均匀且薄的正极电解质界面层。因此,即使在室温下静置 24 小时后,TPDOL@FEC 前体溶液仍能保持液态。组装好的 LiNi 0.6 Co 0.2 Mn 0.2 O 2 ||TPDOL@FEC||Li 电池在 4.4 V (0.5 C) 下循环 100 次后,容量保持率高达 91.5%。这项研究预计将成为追求实际可行的原位形成的 PDOL 电解质的一个飞跃。






























 京公网安备 11010802027423号
京公网安备 11010802027423号