npj Computational Materials ( IF 9.4 ) Pub Date : 2024-09-07 , DOI: 10.1038/s41524-024-01407-2 Brian H. Lee , James P. Larentzos , John K. Brennan , Alejandro Strachan
|
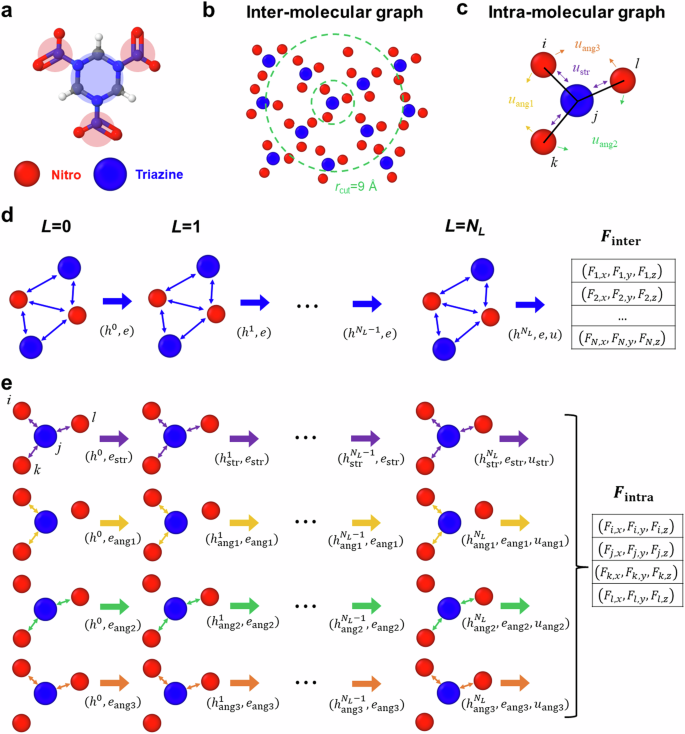
Condense phase molecular systems organize in wide range of distinct molecular configurations, including amorphous melt and glass as well as crystals often exhibiting polymorphism, that originate from their intricate intra- and intermolecular forces. While accurate coarse-grain (CG) models for these materials are critical to understand phenomena beyond the reach of all-atom simulations, current models cannot capture the diversity of molecular structures. We introduce a generally applicable approach to develop CG force fields for molecular crystals combining graph neural networks (GNN) and data from an all-atom simulations and apply it to the high-energy density material RDX. We address the challenge of expanding the training data with relevant configurations via an iterative procedure that performs CG molecular dynamics of processes of interest and reconstructs the atomistic configurations using a pre-trained neural network decoder. The multi-site CG model uses a GNN architecture constructed to satisfy translational invariance and rotational covariance for forces. The resulting model captures both crystalline and amorphous states for a wide range of temperatures and densities.
中文翻译:
分子晶体 RDX 的图神经网络粗粒力场
凝聚相分子系统以各种不同的分子构型组织,包括无定形熔体和玻璃以及经常表现出多晶型现象的晶体,这些多晶型现象源于其复杂的分子内和分子间力。虽然这些材料的准确粗粒 (CG) 模型对于理解全原子模拟无法实现的现象至关重要,但当前模型无法捕捉分子结构的多样性。我们介绍了一种普遍适用的方法,结合图神经网络 (GNN) 和全原子模拟数据来开发分子晶体的 CG 力场,并将其应用于高能量密度材料 RDX。我们通过迭代过程解决了使用相关配置扩展训练数据的挑战,该迭代过程执行感兴趣过程的 CG 分子动力学,并使用预先训练的神经网络解码器重建原子配置。多站点 CG 模型使用 GNN 架构来满足力的平移不变性和旋转协方差。由此产生的模型捕获了各种温度和密度下的晶态和非晶态。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号