Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A high-throughput workflow to analyze sequence-conformation relationships and explore hydrophobic patterning in disordered peptoids
Chem ( IF 19.1 ) Pub Date : 2024-09-06 , DOI: 10.1016/j.chempr.2024.07.025
Erin C. Day , Supraja S. Chittari , Keila C. Cunha , Roy J. Zhao , James N. Dodds , Delaney C. Davis , Erin S. Baker , Rebecca B. Berlow , Joan-Emma Shea , Rishikesh U. Kulkarni , Abigail S. Knight
Chem ( IF 19.1 ) Pub Date : 2024-09-06 , DOI: 10.1016/j.chempr.2024.07.025
Erin C. Day , Supraja S. Chittari , Keila C. Cunha , Roy J. Zhao , James N. Dodds , Delaney C. Davis , Erin S. Baker , Rebecca B. Berlow , Joan-Emma Shea , Rishikesh U. Kulkarni , Abigail S. Knight
|
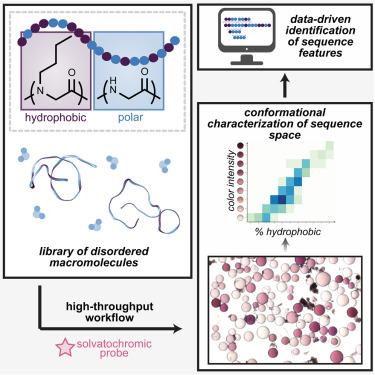
Understanding how a macromolecule’s primary sequence governs its conformational landscape is crucial for elucidating its function, yet these design principles are still emerging for macromolecules with intrinsic disorder. Herein, we introduce a high-throughput workflow that implements a practical colorimetric conformational assay, introduces a semi-automated sequencing protocol using matrix-assisted laser desorption/ionization and tandem mass spectrometry (MALDI-MS/MS), and develops a generalizable sequence-structure algorithm. Using a model system of 20mer peptidomimetics containing polar glycine and hydrophobic N -butylglycine residues, we identified nine classifications of conformational disorder and isolated 122 unique sequences across varied compositions and conformations. Conformational distributions of three compositionally identical library sequences were corroborated through atomistic simulations and ion mobility spectrometry coupled with liquid chromatography. A data-driven strategy was developed using existing sequence variables and data-derived “motifs” to inform a machine-learning algorithm toward conformation prediction. This multifaceted approach enhances our understanding of sequence-conformation relationships and offers a powerful tool for accelerating the discovery of materials with conformational control.
中文翻译:

分析序列-构象关系并探索无序类肽中疏水模式的高通量工作流程
了解大分子的一级序列如何控制其构象景观对于阐明其功能至关重要,但对于具有内在无序的大分子,这些设计原则仍在出现。在此,我们介绍了一种实施实用比色构象测定的高通量工作流程,介绍了一种使用基质辅助激光解吸/电离和串联质谱 (MALDI-MS/MS) 的半自动测序方案,并开发了一种通用的序列结构算法。使用含有极性甘氨酸和疏水性 N-丁基甘氨酸残基的 20 聚体模拟肽模型系统,我们确定了 9 种构象紊乱分类,并分离了不同组成和构象的 122 个独特序列。通过原子模拟和离子迁移谱与液相色谱耦合证实了三个组成相同的文库序列的构象分布。使用现有的序列变量和数据衍生的 “motif” 开发了一种数据驱动的策略,为机器学习算法提供构象预测信息。这种多方面的方法增强了我们对序列-构象关系的理解,并为加速发现具有构象控制的材料提供了强大的工具。
更新日期:2024-09-06
中文翻译:
分析序列-构象关系并探索无序类肽中疏水模式的高通量工作流程
了解大分子的一级序列如何控制其构象景观对于阐明其功能至关重要,但对于具有内在无序的大分子,这些设计原则仍在出现。在此,我们介绍了一种实施实用比色构象测定的高通量工作流程,介绍了一种使用基质辅助激光解吸/电离和串联质谱 (MALDI-MS/MS) 的半自动测序方案,并开发了一种通用的序列结构算法。使用含有极性甘氨酸和疏水性 N-丁基甘氨酸残基的 20 聚体模拟肽模型系统,我们确定了 9 种构象紊乱分类,并分离了不同组成和构象的 122 个独特序列。通过原子模拟和离子迁移谱与液相色谱耦合证实了三个组成相同的文库序列的构象分布。使用现有的序列变量和数据衍生的 “motif” 开发了一种数据驱动的策略,为机器学习算法提供构象预测信息。这种多方面的方法增强了我们对序列-构象关系的理解,并为加速发现具有构象控制的材料提供了强大的工具。






























 京公网安备 11010802027423号
京公网安备 11010802027423号