当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Equation-of-motion orbital-optimized coupled-cluster doubles method with the density-fitting approximation: An efficient implementation
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-09-05 , DOI: 10.1002/jcc.27495 Aslı Ünal 1 , Uğur Bozkaya 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-09-05 , DOI: 10.1002/jcc.27495 Aslı Ünal 1 , Uğur Bozkaya 1
Affiliation
|
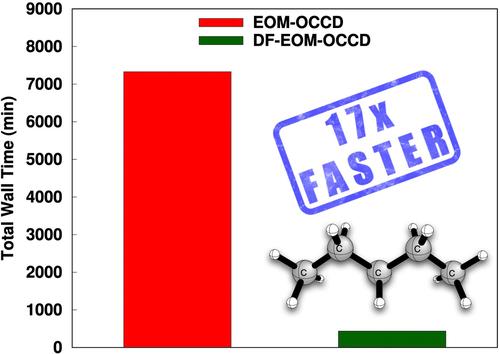
Orbital-optimized coupled-cluster methods are very helpful for theoretical predictions of the molecular properties of challenging chemical systems, such as excited states. In this research, an efficient implementation of the equation-of-motion orbital-optimized coupled-cluster doubles method with the density-fitting (DF) approach, denoted by DF-EOM-OCCD, is presented. The computational cost of the DF-EOM-OCCD method for excitation energies is compared with that of the conventional EOM-OCCD method. Our results demonstrate that DF-EOM-OCCD excitation energies are dramatically accelerated compared to EOM-OCCD. There are almost 17-fold reductions for the
中文翻译:

具有密度拟合近似的运动方程轨道优化耦合簇双精度方法:一种高效的实现
轨道优化的耦合簇方法对于具有挑战性的化学系统(如激发态)的分子性质的理论预测非常有帮助。在这项研究中,提出了一种用密度拟合 (DF) 方法(用 DF-EOM-OCCD 表示)的运动方程轨道优化耦合簇双精度方法的有效实现。将 DF-EOM-OCCD 方法的激发能量计算成本与传统的 EOM-OCCD 方法进行了比较。我们的结果表明,与 EOM-OCCD 相比,DF-EOM-OCCD 激发能量显着加速。C 分子在 aug-cc-pVTZ 基基上与 RHF 参比的还原几乎是 17 倍 。这种显著的性能改进来自于 DF 方法降低的积分变换成本以及对粒子-粒子阶梯 (PPL) 项的有效评估,这是评估成本最高的项。此外,我们的结果表明,DF-EOM-OCCD 方法对于开壳分子系统中激发能的计算非常有帮助。总的来说,我们得出结论,我们的新 DF-EOM-OCCD 实现对于研究大型具有挑战性的化学系统中的激发态非常有前途。
更新日期:2024-09-05
中文翻译:
具有密度拟合近似的运动方程轨道优化耦合簇双精度方法:一种高效的实现
轨道优化的耦合簇方法对于具有挑战性的化学系统(如激发态)的分子性质的理论预测非常有帮助。在这项研究中,提出了一种用密度拟合 (DF) 方法(用 DF-EOM-OCCD 表示)的运动方程轨道优化耦合簇双精度方法的有效实现。将 DF-EOM-OCCD 方法的激发能量计算成本与传统的 EOM-OCCD 方法进行了比较。我们的结果表明,与 EOM-OCCD 相比,DF-EOM-OCCD 激发能量显着加速。C






























 京公网安备 11010802027423号
京公网安备 11010802027423号