当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Chemically accurate predictions for water adsorption on Brønsted sites of zeolite H-MFI
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-09-04 , DOI: 10.1039/d4cp02851a Henning Windeck 1 , Fabian Berger 1 , Joachim Sauer 1, 2
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-09-04 , DOI: 10.1039/d4cp02851a Henning Windeck 1 , Fabian Berger 1 , Joachim Sauer 1, 2
Affiliation
|
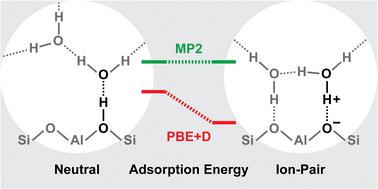
We investigate the adsorption of water molecules in the zeolite H-MFI at isolated Brønsted acid sites (BAS) for loadings of 1, 2, and 3 H2O/BAS. We consider two approaches to the O3Al–O(H)–Si sites: the Brønsted-type approach of H2O to the acidic proton and the Lewis-type approach to the aluminium atom of the AlO4 tetrahedron. From the twelve crystallographically inequivalent framework sites for Al, a representative set of six active site positions is chosen. For them, we calculate CCSD(T)-quality adsorption energies at MP2-quality adsorption structures for different approaches, 48 in total. The Brønsted-type approach is favoured for most cases but the Lewis-type approach has similar stability for some framework positions. We predict heats of adsorption per molecule ranging from 60 to 76, 56 to 65, and 56 to 64 kJ mol−1 for loadings of 1, 2, and 3 H2O/BAS, respectively. For 1 H2O/BAS, the experimental result (70 kJ mol−1) falls into the range of our predictions, whereas for 2 and 3 H2O/BAS, the measured adsorption heats per molecule (74 and 70 kJ mol−1, respectively) are larger than our predictions. For 2 H2O/BAS, the ion-pair structure generated by proton transfer to the water dimer competes with the neutral adsorption complex. The DFT adsorption energies (PBE+D2) deviate significantly from the CCSD(T)-quality reference energies, by up to 25 kJ mol−1 for 1 H2O/BAS, 25 kJ mol−1 per H2O for 2 H2O/BAS, and 18 kJ mol−1 per H2O for 3 H2O/BAS. Specifically, PBE+D2 overstabilises the ion-pair structure, i.e. in many cases the PBE+D2 error is much larger for ionic than for neutral adsorption structures.
中文翻译:

对 H-MFI 沸石布朗斯台德位点上的水吸附进行化学准确预测
我们研究了在 1、2 和 3 H 2 O/BAS 负载量下,H-MFI 沸石中孤立的布朗斯台德酸位点 (BAS) 上水分子的吸附情况。我们考虑O 3 Al-O(H)-Si 位点的两种方法:H 2 O 酸性质子的布朗斯台德型方法和AlO 4四面体铝原子的路易斯型方法。从 Al 的 12 个晶体学不等价的骨架位点中,选择了一组具有代表性的六个活性位点位置。对于它们,我们计算了不同方法在 MP2 质量吸附结构上的 CCSD(T) 质量吸附能,总共 48 种方法。布朗斯台德型方法在大多数情况下受到青睐,但刘易斯型方法对于某些框架位置具有类似的稳定性。我们预测对于 1、2 和 3 H 2 O/BAS 的负载量,每个分子的吸附热分别为 60 至 76、56 至 65 和 56 至 64 kJ mol -1 。对于 1 H 2 O/BAS,实验结果 (70 kJ mol −1 ) 落入我们的预测范围内,而对于 2 和 3 H 2 O/BAS,测量的每个分子的吸附热 (74 和 70 kJ mol − 1 )比我们的预测要大。对于2 H 2 O/BAS,质子转移到水二聚体产生的离子对结构与中性吸附络合物竞争。 DFT 吸附能 (PBE+D2) 与 CCSD(T) 质量参考能的偏差显着,1 H 2 O/BAS 的偏差高达 25 kJ mol -1,2 H 的每 H 2 O 偏差高达 25 kJ mol -1 2 O/BAS,以及对于3 H 2 O/BAS,每H 2 O 18 kJ mol -1 。具体而言,PBE+D2 使离子对结构过度稳定,即,在许多情况下,离子吸附结构的PBE+D2 误差比中性吸附结构的误差大得多。
更新日期:2024-09-05
中文翻译:
对 H-MFI 沸石布朗斯台德位点上的水吸附进行化学准确预测
我们研究了在 1、2 和 3 H 2 O/BAS 负载量下,H-MFI 沸石中孤立的布朗斯台德酸位点 (BAS) 上水分子的吸附情况。我们考虑O 3 Al-O(H)-Si 位点的两种方法:H 2 O 酸性质子的布朗斯台德型方法和AlO 4四面体铝原子的路易斯型方法。从 Al 的 12 个晶体学不等价的骨架位点中,选择了一组具有代表性的六个活性位点位置。对于它们,我们计算了不同方法在 MP2 质量吸附结构上的 CCSD(T) 质量吸附能,总共 48 种方法。布朗斯台德型方法在大多数情况下受到青睐,但刘易斯型方法对于某些框架位置具有类似的稳定性。我们预测对于 1、2 和 3 H 2 O/BAS 的负载量,每个分子的吸附热分别为 60 至 76、56 至 65 和 56 至 64 kJ mol -1 。对于 1 H 2 O/BAS,实验结果 (70 kJ mol −1 ) 落入我们的预测范围内,而对于 2 和 3 H 2 O/BAS,测量的每个分子的吸附热 (74 和 70 kJ mol − 1 )比我们的预测要大。对于2 H 2 O/BAS,质子转移到水二聚体产生的离子对结构与中性吸附络合物竞争。 DFT 吸附能 (PBE+D2) 与 CCSD(T) 质量参考能的偏差显着,1 H 2 O/BAS 的偏差高达 25 kJ mol -1,2 H 的每 H 2 O 偏差高达 25 kJ mol -1 2 O/BAS,以及对于3 H 2 O/BAS,每H 2 O 18 kJ mol -1 。具体而言,PBE+D2 使离子对结构过度稳定,即,在许多情况下,离子吸附结构的PBE+D2 误差比中性吸附结构的误差大得多。
































 京公网安备 11010802027423号
京公网安备 11010802027423号