当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Contact-Map-Driven Exploration of Heterogeneous Protein-Folding Paths
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-04 , DOI: 10.1021/acs.jctc.4c00878 Ziad Fakhoury 1 , Gabriele C Sosso 1 , Scott Habershon 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-04 , DOI: 10.1021/acs.jctc.4c00878 Ziad Fakhoury 1 , Gabriele C Sosso 1 , Scott Habershon 1
Affiliation
|
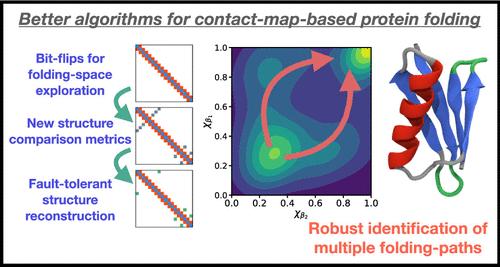
We have recently shown how physically realizable protein-folding pathways can be generated using directed walks in the space of inter-residue contact-maps; combined with a back-transformation to move from protein contact-maps to Cartesian coordinates, we have demonstrated how this approach can generate protein-folding trajectory ensembles without recourse to molecular dynamics. In this article, we demonstrate that this framework can be used to study a challenging protein-folding problem that is known to exhibit two different folding paths which were previously identified through molecular dynamics simulation at several different temperatures. From the viewpoint of protein-folding mechanism prediction, this particular problem is extremely challenging to address, specifically involving folding to an identical nontrivial compact native structure along distinct pathways defined by heterogeneous secondary structural elements. Here, we show how our previously reported contact-map-based protein-folding strategy can be significantly enhanced to enable accurate and robust prediction of heterogeneous folding paths by (i) introducing a novel topologically informed metric for comparing two protein contact maps, (ii) reformulating our graph-represented folding path generation, and (iii) introducing a new and more reliable structural back-mapping algorithm. These changes improve the reliability of generating structurally sound folding intermediates and dramatically decrease the number of physically irrelevant folding intermediates generated by our previous simulation strategy. Most importantly, we demonstrate how our enhanced folding algorithm can successfully identify the alternative folding mechanisms of a multifolding-pathway protein, in line with direct molecular dynamics simulations.
中文翻译:

接触图驱动的异质蛋白质折叠路径探索
我们最近展示了如何使用残基间接触图空间中的定向行走来生成物理上可实现的蛋白质折叠途径;结合从蛋白质接触图到笛卡尔坐标的反向转换,我们已经证明了这种方法如何能够在不依赖分子动力学的情况下生成蛋白质折叠轨迹集合。在本文中,我们证明该框架可用于研究具有挑战性的蛋白质折叠问题,已知该问题表现出两种不同的折叠路径,这两种折叠路径先前是通过几种不同温度下的分子动力学模拟确定的。从蛋白质折叠机制预测的角度来看,解决这个特殊问题极具挑战性,特别是涉及沿着异质二级结构元件定义的不同途径折叠成相同的非平凡紧凑的天然结构。在这里,我们展示了如何显着增强我们之前报道的基于接触图的蛋白质折叠策略,以通过(i)引入一种新的拓扑信息度量来比较两个蛋白质接触图,从而能够准确而稳健地预测异质折叠路径,(ii) )重新制定我们的图形表示的折叠路径生成,以及(iii)引入一种新的、更可靠的结构反向映射算法。这些变化提高了生成结构合理的折叠中间体的可靠性,并显着减少了我们之前的模拟策略生成的物理上不相关的折叠中间体的数量。 最重要的是,我们展示了我们的增强折叠算法如何能够根据直接分子动力学模拟成功识别多重折叠途径蛋白质的替代折叠机制。
更新日期:2024-09-04
中文翻译:
接触图驱动的异质蛋白质折叠路径探索
我们最近展示了如何使用残基间接触图空间中的定向行走来生成物理上可实现的蛋白质折叠途径;结合从蛋白质接触图到笛卡尔坐标的反向转换,我们已经证明了这种方法如何能够在不依赖分子动力学的情况下生成蛋白质折叠轨迹集合。在本文中,我们证明该框架可用于研究具有挑战性的蛋白质折叠问题,已知该问题表现出两种不同的折叠路径,这两种折叠路径先前是通过几种不同温度下的分子动力学模拟确定的。从蛋白质折叠机制预测的角度来看,解决这个特殊问题极具挑战性,特别是涉及沿着异质二级结构元件定义的不同途径折叠成相同的非平凡紧凑的天然结构。在这里,我们展示了如何显着增强我们之前报道的基于接触图的蛋白质折叠策略,以通过(i)引入一种新的拓扑信息度量来比较两个蛋白质接触图,从而能够准确而稳健地预测异质折叠路径,(ii) )重新制定我们的图形表示的折叠路径生成,以及(iii)引入一种新的、更可靠的结构反向映射算法。这些变化提高了生成结构合理的折叠中间体的可靠性,并显着减少了我们之前的模拟策略生成的物理上不相关的折叠中间体的数量。 最重要的是,我们展示了我们的增强折叠算法如何能够根据直接分子动力学模拟成功识别多重折叠途径蛋白质的替代折叠机制。






























 京公网安备 11010802027423号
京公网安备 11010802027423号