当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Simulating Bacterial Membrane Models at the Atomistic Level: A Force Field Comparison
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-03 , DOI: 10.1021/acs.jctc.4c00204 Alexandre Blanco-González 1, 2, 3 , Anika Wurl 4 , Tiago Mendes Ferreira 4 , Ángel Piñeiro 1 , Rebeca Garcia-Fandino 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-03 , DOI: 10.1021/acs.jctc.4c00204 Alexandre Blanco-González 1, 2, 3 , Anika Wurl 4 , Tiago Mendes Ferreira 4 , Ángel Piñeiro 1 , Rebeca Garcia-Fandino 2
Affiliation
|
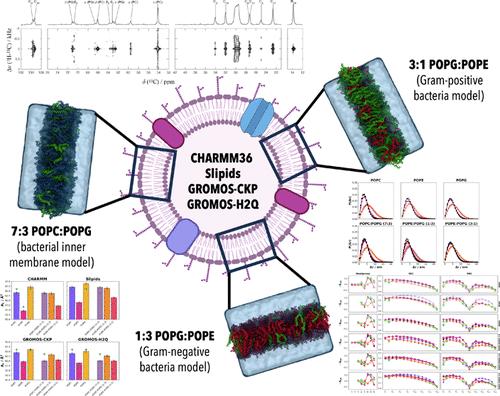
Molecular dynamics (MD) simulations are currently an indispensable tool to understand both the dynamic and nanoscale organization of cell membrane models. A large number of quantitative parameters can be extracted from these simulations, but their reliability is determined by the quality of the employed force field and the simulation parameters. Much of the work on parametrizing and optimizing force fields for biomembrane modeling has been focused on homogeneous bilayers with a single phospholipid type. However, these may not perform effectively or could even be unsuitable for lipid mixtures commonly employed in membrane models. This work aims to fill this gap by comparing MD simulation results of several bacterial membrane models using different force fields and simulation parameters, namely, CHARMM36, Slipids, and GROMOS-CKP. Furthermore, the hydrogen isotope exchange (HIE) method, combined with GROMOS-CKP (GROMOS-H2Q), was also tested to check for the impact of this acceleration strategy on the performance of the force field. A common set of simulation parameters was employed for all of the force fields in addition to those corresponding to the original parametrization of each of them. Furthermore, new experimental order parameter values determined from NMR of several lipid mixtures are also reported to compare them with those determined from MD simulations. Our results reveal that most of the calculated physical properties of bacterial membrane models from MD simulations are substantially force field and lipid composition dependent. Some lipid mixtures exhibit nearly ideal behaviors, while the interaction of different lipid types in other mixtures is highly synergistic. None of the employed force fields seem to be clearly superior to the other three, each having its own strengths and weaknesses. Slipids are notably effective at replicating the order parameters for all acyl chains, including those in lipid mixtures, but they offer the least accurate results for headgroup parameters. Conversely, CHARMM provides almost perfect estimates for the order parameters of the headgroups but tends to overestimate those of the lipid tails. The GROMOS parametrizations deliver reasonable order parameters for entire lipid molecules, including multicomponent bilayers, although they do not reach the accuracy of Slipids for tails or CHARMM for headgroups. Importantly, GROMOS-H2Q stands out for its computational efficiency, being at least 3 times faster than GROMOS, which is already faster than both CHARMM and Slipids. In turn, GROMOS-H2Q yields much higher compressibilities compared to all other parametrizations.
中文翻译:

在原子水平上模拟细菌膜模型:力场比较
分子动力学 (MD) 模拟目前是了解细胞膜模型的动态和纳米级组织不可或缺的工具。从这些模拟中可以提取大量定量参数,但它们的可靠性取决于所采用的力场和模拟参数的质量。生物膜建模的参数化和优化力场的大部分工作都集中在具有单一磷脂类型的均质双层上。然而,这些可能无法有效发挥作用,甚至可能不适合膜模型中常用的脂质混合物。本工作旨在通过比较使用不同力场和模拟参数的几种细菌膜模型(即 CHARMM36、Slipids 和 GROMOS-CKP)的 MD 模拟结果来填补这一空白。此外,还测试了氢同位素交换(HIE)方法与GROMOS-CKP(GROMOS-H2Q)的结合,以检查这种加速策略对力场性能的影响。除了与每个力场的原始参数化相对应的参数之外,所有力场还采用了一组通用的模拟参数。此外,还报告了从几种脂质混合物的 NMR 确定的新实验顺序参数值,以将它们与 MD 模拟确定的值进行比较。我们的结果表明,MD 模拟计算出的细菌膜模型的大部分物理特性基本上依赖于力场和脂质成分。一些脂质混合物表现出近乎理想的行为,而其他混合物中不同脂质类型的相互作用具有高度协同作用。 所使用的力场似乎没有一个明显优于其他三个,每个力场都有自己的优点和缺点。滑脂在复制所有酰基链(包括脂质混合物中的酰基链)的有序参数方面尤其有效,但它们提供的头基参数结果最不准确。相反,CHARMM 对头基的有序参数提供了近乎完美的估计,但往往高估了脂质尾部的有序参数。 GROMOS 参数化为整个脂质分子(包括多组分双层)提供了合理的有序参数,尽管它们没有达到尾部 Slipids 或头基 CHARMM 的精度。重要的是,GROMOS-H2Q 因其计算效率而脱颖而出,比 GROMOS 快至少 3 倍,而 GROMOS 已经比 CHARMM 和 Slipids 都快。反过来,与所有其他参数化相比,GROMOS-H2Q 产生更高的压缩率。
更新日期:2024-09-03
中文翻译:
在原子水平上模拟细菌膜模型:力场比较
分子动力学 (MD) 模拟目前是了解细胞膜模型的动态和纳米级组织不可或缺的工具。从这些模拟中可以提取大量定量参数,但它们的可靠性取决于所采用的力场和模拟参数的质量。生物膜建模的参数化和优化力场的大部分工作都集中在具有单一磷脂类型的均质双层上。然而,这些可能无法有效发挥作用,甚至可能不适合膜模型中常用的脂质混合物。本工作旨在通过比较使用不同力场和模拟参数的几种细菌膜模型(即 CHARMM36、Slipids 和 GROMOS-CKP)的 MD 模拟结果来填补这一空白。此外,还测试了氢同位素交换(HIE)方法与GROMOS-CKP(GROMOS-H2Q)的结合,以检查这种加速策略对力场性能的影响。除了与每个力场的原始参数化相对应的参数之外,所有力场还采用了一组通用的模拟参数。此外,还报告了从几种脂质混合物的 NMR 确定的新实验顺序参数值,以将它们与 MD 模拟确定的值进行比较。我们的结果表明,MD 模拟计算出的细菌膜模型的大部分物理特性基本上依赖于力场和脂质成分。一些脂质混合物表现出近乎理想的行为,而其他混合物中不同脂质类型的相互作用具有高度协同作用。 所使用的力场似乎没有一个明显优于其他三个,每个力场都有自己的优点和缺点。滑脂在复制所有酰基链(包括脂质混合物中的酰基链)的有序参数方面尤其有效,但它们提供的头基参数结果最不准确。相反,CHARMM 对头基的有序参数提供了近乎完美的估计,但往往高估了脂质尾部的有序参数。 GROMOS 参数化为整个脂质分子(包括多组分双层)提供了合理的有序参数,尽管它们没有达到尾部 Slipids 或头基 CHARMM 的精度。重要的是,GROMOS-H2Q 因其计算效率而脱颖而出,比 GROMOS 快至少 3 倍,而 GROMOS 已经比 CHARMM 和 Slipids 都快。反过来,与所有其他参数化相比,GROMOS-H2Q 产生更高的压缩率。






























 京公网安备 11010802027423号
京公网安备 11010802027423号