当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Optimal Molecular Design: Generative Active Learning Combining REINVENT with Precise Binding Free Energy Ranking Simulations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-03 , DOI: 10.1021/acs.jctc.4c00576 Hannes H Loeffler 1 , Shunzhou Wan 2 , Marco Klähn 1 , Agastya P Bhati 2 , Peter V Coveney 2, 3, 4
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-09-03 , DOI: 10.1021/acs.jctc.4c00576 Hannes H Loeffler 1 , Shunzhou Wan 2 , Marco Klähn 1 , Agastya P Bhati 2 , Peter V Coveney 2, 3, 4
Affiliation
|
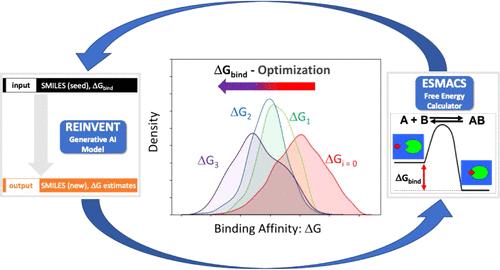
Active learning (AL) is a specific instance of sequential experimental design and uses machine learning to intelligently choose the next data point or batch of molecular structures to be evaluated. In this sense, it closely mimics the iterative design-make-test-analysis cycle of laboratory experiments to find optimized compounds for a given design task. Here, we describe an AL protocol which combines generative molecular AI, using REINVENT, and physics-based absolute binding free energy molecular dynamics simulation, using ESMACS, to discover new ligands for two different target proteins, 3CLpro and TNKS2. We have deployed our generative active learning (GAL) protocol on Frontier, the world’s only exa-scale machine. We show that the protocol can find higher-scoring molecules compared to the baseline, a surrogate ML docking model for 3CLpro and compounds with experimentally determined binding affinities for TNKS2. The ligands found are also chemically diverse and occupy a different chemical space than the baseline. We vary the batch sizes that are put forward for free energy assessment in each GAL cycle to assess the impact on their efficiency on the GAL protocol and recommend their optimal values in different scenarios. Overall, we demonstrate a powerful capability of the combination of physics-based and AI methods which yields effective chemical space sampling at an unprecedented scale and is of immediate and direct relevance to modern, data-driven drug discovery.
中文翻译:

最优分子设计:将 REINVENT 与精确的结合自由能排序模拟相结合的生成主动学习
主动学习(AL)是顺序实验设计的一个具体实例,它使用机器学习来智能地选择下一个数据点或要评估的一批分子结构。从这个意义上说,它密切模仿实验室实验的迭代设计-制造-测试-分析循环,为给定的设计任务找到优化的化合物。在这里,我们描述了一种 AL 协议,该协议结合了使用 REINVENT 的生成分子 AI 和使用 ESMACS 的基于物理的绝对结合自由能分子动力学模拟,以发现两种不同目标蛋白 3CL pro和 TNKS2 的新配体。我们已经在世界上唯一的百亿亿级机器 Frontier 上部署了生成主动学习 (GAL) 协议。我们表明,与基线相比,该方案可以找到得分更高的分子、3CL pro的替代 ML 对接模型以及通过实验确定的 TNKS2 结合亲和力的化合物。发现的配体在化学上也具有多样性,并且占据与基线不同的化学空间。我们改变每个 GAL 周期中为免费能源评估提出的批量大小,以评估其对 GAL 协议效率的影响,并在不同场景下推荐其最佳值。总的来说,我们展示了基于物理和人工智能方法相结合的强大能力,可以以前所未有的规模产生有效的化学空间采样,并且与现代数据驱动的药物发现直接相关。
更新日期:2024-09-03
中文翻译:
最优分子设计:将 REINVENT 与精确的结合自由能排序模拟相结合的生成主动学习
主动学习(AL)是顺序实验设计的一个具体实例,它使用机器学习来智能地选择下一个数据点或要评估的一批分子结构。从这个意义上说,它密切模仿实验室实验的迭代设计-制造-测试-分析循环,为给定的设计任务找到优化的化合物。在这里,我们描述了一种 AL 协议,该协议结合了使用 REINVENT 的生成分子 AI 和使用 ESMACS 的基于物理的绝对结合自由能分子动力学模拟,以发现两种不同目标蛋白 3CL pro和 TNKS2 的新配体。我们已经在世界上唯一的百亿亿级机器 Frontier 上部署了生成主动学习 (GAL) 协议。我们表明,与基线相比,该方案可以找到得分更高的分子、3CL pro的替代 ML 对接模型以及通过实验确定的 TNKS2 结合亲和力的化合物。发现的配体在化学上也具有多样性,并且占据与基线不同的化学空间。我们改变每个 GAL 周期中为免费能源评估提出的批量大小,以评估其对 GAL 协议效率的影响,并在不同场景下推荐其最佳值。总的来说,我们展示了基于物理和人工智能方法相结合的强大能力,可以以前所未有的规模产生有效的化学空间采样,并且与现代数据驱动的药物发现直接相关。






























 京公网安备 11010802027423号
京公网安备 11010802027423号