当前位置:
X-MOL 学术
›
J. Mater. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
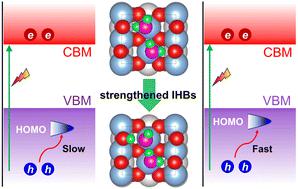
Enhancement of hole capture and water dissociation on rutile TiO2(110) by intermolecular hydrogen bonding: time-domain ab initio study
Journal of Materials Chemistry A ( IF 10.7 ) Pub Date : 2024-09-02 , DOI: 10.1039/d4ta04750h Yitong Zhang , Cheng Cheng , Yifan Wu , Oleg V Prezhdo , Run Long
Journal of Materials Chemistry A ( IF 10.7 ) Pub Date : 2024-09-02 , DOI: 10.1039/d4ta04750h Yitong Zhang , Cheng Cheng , Yifan Wu , Oleg V Prezhdo , Run Long
|
Photocatalytic water splitting has been a focal point of research to solve energy and environmental issues. However, the understanding of photocatalytic water splitting and coupled dynamics of photogenerated charge carriers at molecule/semiconductor interfaces is still limited. We have combined ab initio molecular dynamics, real-time time-dependent density functional theory, and nonadiabatic molecular dynamics to study the dissociation of water and capture of photogenerated holes on the pristine rutile TiO2(110) surface. Our simulations indicate that intermolecular hydrogen bonding (IHB) between water molecules facilitates water dissociation. The dissociation energy of water molecules in a pristine, non-dissociated structure is reduced by 15%, from 0.26 eV to 0.21 eV, due to IHB. In the semi-dissociated structure, the dissociation energy of a water molecule is only 0.13 eV, owing to proton transfer induced by IHB. In the semi-dissociated structure, IHB between H2O and terminal hydroxyl (OtH) stabilizes the dissociated structure. Furthermore, IHB promotes spatial isolation of OtH and bridging hydroxyl (ObrH) and inhibits their recombination. The stabilized dissociated structure activates high-frequency vibrational modes that increase the nonadiabatic coupling and promote hole capture on a femtosecond timescale, accelerating the capture rate by 36%. The findings provide important insights into photo-dissociation of water on rutile TiO2(110), particularly shedding light on the impact of key intermediates on the photocatalytic process.
中文翻译:

通过分子间氢键增强金红石 TiO2(110) 上的空穴捕获和水解离:时域从头开始研究
光催化水分解一直是解决能源和环境问题的研究焦点。然而,对光催化水分解和分子/半导体界面光生载流子耦合动力学的理解仍然有限。我们结合从头算分子动力学、实时依赖时间的密度泛函理论和非绝热分子动力学来研究水的解离和原始金红石TiO 2 (110) 表面上光生空穴的捕获。我们的模拟表明,水分子之间的分子间氢键(IHB)有利于水的解离。由于 IHB,原始非解离结构中水分子的解离能降低了 15%,从 0.26 eV 降至 0.21 eV。在半解离结构中,由于 IHB 诱导的质子转移,水分子的解离能仅为 0.13 eV。在半解离结构中,H 2 O和末端羟基(O t H)之间的IHB稳定了解离结构。此外,IHB 促进 O t H 和桥接羟基 (O br H) 的空间隔离并抑制它们的重组。稳定的解离结构激活高频振动模式,增加非绝热耦合并促进飞秒时间尺度上的空穴捕获,将捕获率加快36%。 这些发现为水在金红石 TiO 2 (110) 上的光解提供了重要的见解,特别是揭示了关键中间体对光催化过程的影响。
更新日期:2024-09-02
中文翻译:
通过分子间氢键增强金红石 TiO2(110) 上的空穴捕获和水解离:时域从头开始研究
光催化水分解一直是解决能源和环境问题的研究焦点。然而,对光催化水分解和分子/半导体界面光生载流子耦合动力学的理解仍然有限。我们结合从头算分子动力学、实时依赖时间的密度泛函理论和非绝热分子动力学来研究水的解离和原始金红石TiO 2 (110) 表面上光生空穴的捕获。我们的模拟表明,水分子之间的分子间氢键(IHB)有利于水的解离。由于 IHB,原始非解离结构中水分子的解离能降低了 15%,从 0.26 eV 降至 0.21 eV。在半解离结构中,由于 IHB 诱导的质子转移,水分子的解离能仅为 0.13 eV。在半解离结构中,H 2 O和末端羟基(O t H)之间的IHB稳定了解离结构。此外,IHB 促进 O t H 和桥接羟基 (O br H) 的空间隔离并抑制它们的重组。稳定的解离结构激活高频振动模式,增加非绝热耦合并促进飞秒时间尺度上的空穴捕获,将捕获率加快36%。 这些发现为水在金红石 TiO 2 (110) 上的光解提供了重要的见解,特别是揭示了关键中间体对光催化过程的影响。






























 京公网安备 11010802027423号
京公网安备 11010802027423号