当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Systematic Improvement of Quantum Monte Carlo Calculations in Transition Metal Oxides: sCI-Driven Wavefunction Optimization for Reliable Band Gap Prediction
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-30 , DOI: 10.1021/acs.jctc.4c00335 Hyeondeok Shin 1 , Kevin Gasperich 1 , Tomas Rojas 2, 3 , Anh T Ngo 2, 3 , Jaron T Krogel 4 , Anouar Benali 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-30 , DOI: 10.1021/acs.jctc.4c00335 Hyeondeok Shin 1 , Kevin Gasperich 1 , Tomas Rojas 2, 3 , Anh T Ngo 2, 3 , Jaron T Krogel 4 , Anouar Benali 1
Affiliation
|
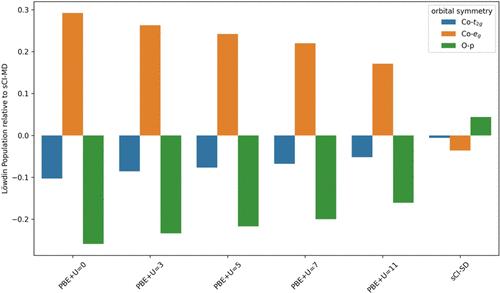
Accurate determination of the electronic properties of correlated oxides remains a significant challenge for computational theory. Traditional Hubbard-corrected density functional theory (DFT+U) frequently encounters limitations in precisely capturing electron correlation, particularly in predicting band gaps. We introduce a systematic methodology to enhance the accuracy of diffusion Monte Carlo (DMC) simulations for both ground and excited states, focusing on LiCoO2 as a case study. By employing a selected configuration interaction (sCI) approach, we demonstrate the capability to optimize wavefunctions beyond the constraints of single-reference DFT+U trial wavefunctions. We show that the sCI framework enables accurate prediction of band gaps in LiCoO2, closely aligning with experimental values and substantially improving traditional computational methods. The study uncovers a nuanced mixed state of t2g and eg orbitals at the band edges that is not captured by conventional single-reference methods, further elucidating the limitations of PBE+U in describing d–d excitations. Our findings advocate for the adoption of beyond-DFT methodologies, such as sCI, to capture the essential physics of excited-state wavefunctions in strongly correlated materials. The improved accuracy in band gap predictions and the ability to generate more reliable trial wavefunctions for DMC calculations underscore the potential of this approach for broader applications in the study of correlated oxides. This work not only provides a pathway for more accurate simulations of electronic structures in complex materials but also suggests a framework for future investigations of the excited states of other challenging systems.
中文翻译:

过渡金属氧化物中量子蒙特卡罗计算的系统改进:sCI 驱动的波函数优化以实现可靠的带隙预测
准确确定相关氧化物的电子特性仍然是计算理论的重大挑战。传统的哈伯德校正密度泛函理论 (DFT+U) 在精确捕获电子相关性方面经常遇到限制,特别是在预测带隙方面。我们引入了一种系统方法来提高基态和激发态扩散蒙特卡罗 (DMC) 模拟的准确性,重点以 LiCoO 2作为案例研究。通过采用选定配置交互 (sCI) 方法,我们展示了超越单参考 DFT+U 试验波函数的限制来优化波函数的能力。我们表明,sCI 框架能够准确预测 LiCoO 2中的带隙,与实验值紧密结合,并显着改进传统的计算方法。该研究揭示了能带边缘处 t 2g和 e g轨道的微妙混合态,这是传统单参考方法无法捕获的,进一步阐明了 PBE+U 在描述 d-d 激发方面的局限性。我们的研究结果主张采用超越 DFT 的方法(例如 sCI)来捕获强相关材料中激发态波函数的基本物理原理。带隙预测精度的提高以及为 DMC 计算生成更可靠的试验波函数的能力强调了这种方法在相关氧化物研究中更广泛应用的潜力。这项工作不仅为复杂材料中电子结构的更准确模拟提供了一条途径,而且还为未来研究其他具有挑战性的系统的激发态提供了一个框架。
更新日期:2024-08-30
中文翻译:
过渡金属氧化物中量子蒙特卡罗计算的系统改进:sCI 驱动的波函数优化以实现可靠的带隙预测
准确确定相关氧化物的电子特性仍然是计算理论的重大挑战。传统的哈伯德校正密度泛函理论 (DFT+U) 在精确捕获电子相关性方面经常遇到限制,特别是在预测带隙方面。我们引入了一种系统方法来提高基态和激发态扩散蒙特卡罗 (DMC) 模拟的准确性,重点以 LiCoO 2作为案例研究。通过采用选定配置交互 (sCI) 方法,我们展示了超越单参考 DFT+U 试验波函数的限制来优化波函数的能力。我们表明,sCI 框架能够准确预测 LiCoO 2中的带隙,与实验值紧密结合,并显着改进传统的计算方法。该研究揭示了能带边缘处 t 2g和 e g轨道的微妙混合态,这是传统单参考方法无法捕获的,进一步阐明了 PBE+U 在描述 d-d 激发方面的局限性。我们的研究结果主张采用超越 DFT 的方法(例如 sCI)来捕获强相关材料中激发态波函数的基本物理原理。带隙预测精度的提高以及为 DMC 计算生成更可靠的试验波函数的能力强调了这种方法在相关氧化物研究中更广泛应用的潜力。这项工作不仅为复杂材料中电子结构的更准确模拟提供了一条途径,而且还为未来研究其他具有挑战性的系统的激发态提供了一个框架。






























 京公网安备 11010802027423号
京公网安备 11010802027423号