当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine-Learning-Accelerated DFT Conformal Sampling of Catalytic Processes
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-30 , DOI: 10.1021/acs.jctc.4c00643 Thantip Roongcharoen 1 , Giorgio Conter 1, 2 , Luca Sementa 3 , Giacomo Melani 1 , Alessandro Fortunelli 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-30 , DOI: 10.1021/acs.jctc.4c00643 Thantip Roongcharoen 1 , Giorgio Conter 1, 2 , Luca Sementa 3 , Giacomo Melani 1 , Alessandro Fortunelli 1
Affiliation
|
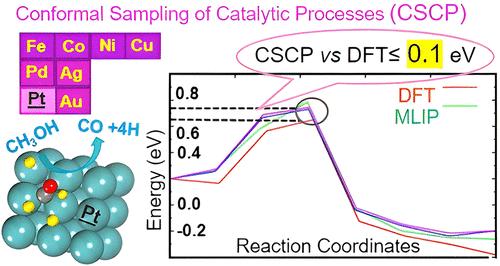
Computational modeling of catalytic processes at gas/solid interfaces plays an increasingly important role in chemistry, enabling accelerated materials and process optimization and rational design. However, efficiency, accuracy, thoroughness, and throughput must be enhanced to maximize its practical impact. By combining interpolation of DFT energetics via highly accurate Machine-Learning Potentials with conformal techniques for building the training database, we present here an original approach (that we name Conformal Sampling of Catalytic Processes, CSCP), to accelerate and achieve an accurate and thorough sampling of novel systems by exporting existing information on a worked-out case. We use methanol decomposition (of interest in the field of hydrogen production and storage) as a test catalytic reaction. Starting from worked-out Pt-based systems, we show that after only two iterations of active-learning CSCP is able to provide reaction energy diagrams for a set of 7 diverse systems (Pd, Ni, Au, Ag, Cu, Co, Fe) leading to DFT-accuracy-level predictions. Cases exhibiting a change in adsorption sites and mechanisms are also successfully reproduced as tests of catalytic path modification. The CSCP approach thus offers itself as an operative tool to fully take advantage of accumulated information to achieve high-throughput sampling of catalytic processes.
中文翻译:

机器学习加速的催化过程 DFT 保形采样
气/固界面催化过程的计算建模在化学中发挥着越来越重要的作用,可以加速材料和过程优化以及合理设计。但是,必须提高效率、准确性、彻底性和吞吐量,以最大限度地发挥其实际影响。通过将通过高精度机器学习电位对 DFT 能量学进行插值与构建训练数据库的保形技术相结合,我们在这里提出了一种原始方法(我们称之为催化过程的保形采样,CSCP),通过导出现有信息来加速和实现对新系统的准确和彻底的采样一个工作案例。我们使用甲醇分解(在氢气生产和储存领域感兴趣)作为测试催化反应。从经过开发的基于 Pt 的系统开始,我们表明,仅经过两次主动学习迭代后,CSCP 就能够为一组 7 个不同系统(Pd、Ni、Au、Ag、Cu、Co、Fe)提供反应能图,从而实现 DFT 精度水平的预测。表现出吸附位点和机制变化的情况也成功地再现为催化路径修饰的测试。因此,CSCP 方法将自己作为一种操作工具,以充分利用积累的信息来实现催化过程的高通量采样。
更新日期:2024-08-30
中文翻译:
机器学习加速的催化过程 DFT 保形采样
气/固界面催化过程的计算建模在化学中发挥着越来越重要的作用,可以加速材料和过程优化以及合理设计。但是,必须提高效率、准确性、彻底性和吞吐量,以最大限度地发挥其实际影响。通过将通过高精度机器学习电位对 DFT 能量学进行插值与构建训练数据库的保形技术相结合,我们在这里提出了一种原始方法(我们称之为催化过程的保形采样,CSCP),通过导出现有信息来加速和实现对新系统的准确和彻底的采样一个工作案例。我们使用甲醇分解(在氢气生产和储存领域感兴趣)作为测试催化反应。从经过开发的基于 Pt 的系统开始,我们表明,仅经过两次主动学习迭代后,CSCP 就能够为一组 7 个不同系统(Pd、Ni、Au、Ag、Cu、Co、Fe)提供反应能图,从而实现 DFT 精度水平的预测。表现出吸附位点和机制变化的情况也成功地再现为催化路径修饰的测试。因此,CSCP 方法将自己作为一种操作工具,以充分利用积累的信息来实现催化过程的高通量采样。






























 京公网安备 11010802027423号
京公网安备 11010802027423号