当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Excited state electronic structure of dimethyl disulfide involved in photodissociation at ∼200 nm
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-08-30 , DOI: 10.1039/d4cp02505a Varun Rishi 1 , Neil C Cole-Filipiak 1 , Krupa Ramasesha 1 , Laura M McCaslin 1
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-08-30 , DOI: 10.1039/d4cp02505a Varun Rishi 1 , Neil C Cole-Filipiak 1 , Krupa Ramasesha 1 , Laura M McCaslin 1
Affiliation
|
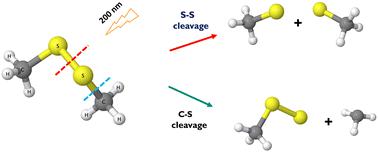
Dimethyl disulfide (DMDS), one of the smallest organic molecules with an S–S bond, serves as a model system for understanding photofragmentation in polypeptides and proteins. Prior studies of DMDS photodissociation excited at ∼266 nm and ∼248 nm have elucidated the mechanisms of S–S and C–S bond cleavage, which involve the lowest excited electronic states S1 and S2. Far less is known about the dissociation mechanisms and electronic structure of relevant excited states of DMDS excited at ∼200 nm. Herein we present calculations of the electronic structure and properties of electronic states S1–S6 accessed when DMDS is excited at ∼200 nm. Our analysis includes a comparison of theoretical and experimental UV spectra, as well as theoretically predicted one-dimensional cuts through the singlet and triplet potential energy surfaces along the S–S and C–S bond dissociation coordinates. Finally, we present calculations of spin–orbit coupling constants at the Franck–Condon geometry to assess the likelihood of ultrafast intersystem crossing. We show that choosing an accurate yet computationally efficient electronic structure method for calculating the S0–S6 potential energy surfaces along relevant dissociation coordinates is challenging due to excited states with doubly excited character and/or mixed Rydberg-valence character. Our findings demonstrate that the extended multi-state complete active space second-order perturbation theory (XMS-CASPT2) balances this computational efficiency and accuracy, as it captures both the Rydberg character of states in the Franck–Condon region and multiconfigurational character toward the bond-dissociation limits. We compare the performance of XMS-CASPT2 to a new variant of equation of motion coupled cluster theory with single, double, and perturbative triple corrections, EOM-CCSD(T)(a)*, finding that EOM-CCSD(T)(a)* significantly improves the treatment of doubly excited states compared to EOM-CCSD, but struggles to quantitatively capture asymptotic energies along bond dissociation coordinates for these states.
中文翻译:

参与~200 nm光解离的二甲基二硫醚的激发态电子结构
二甲基二硫 (DMDS) 是具有 S-S 键的最小有机分子之一,可作为了解多肽和蛋白质光断裂的模型系统。先前对在~266 nm和~248 nm激发的DMDS光解离的研究已经阐明了S-S和C-S键断裂的机制,其中涉及最低激发电子态S 1和S 2 。关于 DMDS 在~200 nm 激发的相关激发态的解离机制和电子结构知之甚少。在此,我们提出了当 DMDS 在 ~200 nm 激发时获得的电子结构和电子态 S 1 –S 6的性质的计算。我们的分析包括理论和实验紫外光谱的比较,以及理论上预测的沿 S-S 和 C-S 键解离坐标穿过单线态和三线态势能表面的一维切割。最后,我们提出了弗兰克-康登几何中自旋轨道耦合常数的计算,以评估超快系间穿越的可能性。我们表明,由于具有双激发特性和/或混合里德伯价特性的激发态,选择准确且计算高效的电子结构方法来计算沿相关解离坐标的 S 0 –S 6势能面是具有挑战性的。 我们的研究结果表明,扩展多态完全主动空间二阶微扰理论 (XMS-CASPT2) 平衡了这种计算效率和准确性,因为它捕获了 Franck-Condon 区域中态的里德堡特征和键的多构型特征-解离限制。我们将 XMS-CASPT2 的性能与具有单、双和微扰三重校正的运动方程耦合簇理论的新变体 EOM-CCSD(T)(a)* 进行比较,发现 EOM-CCSD(T)(a )* 与 EOM-CCSD 相比,显着改善了双激发态的处理,但很难定量捕获这些态沿键解离坐标的渐近能量。
更新日期:2024-08-30
中文翻译:
参与~200 nm光解离的二甲基二硫醚的激发态电子结构
二甲基二硫 (DMDS) 是具有 S-S 键的最小有机分子之一,可作为了解多肽和蛋白质光断裂的模型系统。先前对在~266 nm和~248 nm激发的DMDS光解离的研究已经阐明了S-S和C-S键断裂的机制,其中涉及最低激发电子态S 1和S 2 。关于 DMDS 在~200 nm 激发的相关激发态的解离机制和电子结构知之甚少。在此,我们提出了当 DMDS 在 ~200 nm 激发时获得的电子结构和电子态 S 1 –S 6的性质的计算。我们的分析包括理论和实验紫外光谱的比较,以及理论上预测的沿 S-S 和 C-S 键解离坐标穿过单线态和三线态势能表面的一维切割。最后,我们提出了弗兰克-康登几何中自旋轨道耦合常数的计算,以评估超快系间穿越的可能性。我们表明,由于具有双激发特性和/或混合里德伯价特性的激发态,选择准确且计算高效的电子结构方法来计算沿相关解离坐标的 S 0 –S 6势能面是具有挑战性的。 我们的研究结果表明,扩展多态完全主动空间二阶微扰理论 (XMS-CASPT2) 平衡了这种计算效率和准确性,因为它捕获了 Franck-Condon 区域中态的里德堡特征和键的多构型特征-解离限制。我们将 XMS-CASPT2 的性能与具有单、双和微扰三重校正的运动方程耦合簇理论的新变体 EOM-CCSD(T)(a)* 进行比较,发现 EOM-CCSD(T)(a )* 与 EOM-CCSD 相比,显着改善了双激发态的处理,但很难定量捕获这些态沿键解离坐标的渐近能量。






























 京公网安备 11010802027423号
京公网安备 11010802027423号