当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Investigations and Comparisons of the Amorphous Structures of Adamantane-like Cluster Materials Utilizing Molecular Dynamics Simulations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-29 , DOI: 10.1021/acs.jctc.4c00196 Sebastian Schwan 1, 2 , Benjamin D Klee 3, 4 , Niklas Rinn 5 , Peter R Schreiner 2, 6 , Stefanie Dehnen 5 , Wolf-Christian Pilgrim 3 , Doreen Mollenhauer 1, 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-29 , DOI: 10.1021/acs.jctc.4c00196 Sebastian Schwan 1, 2 , Benjamin D Klee 3, 4 , Niklas Rinn 5 , Peter R Schreiner 2, 6 , Stefanie Dehnen 5 , Wolf-Christian Pilgrim 3 , Doreen Mollenhauer 1, 2
Affiliation
|
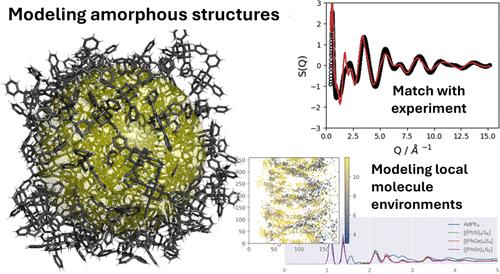
Cluster materials of the composition AdR4 (Ad = adamantane, R = organic substituent) and [(RT)4E6] (R = organic substituent; T = Si, Ge, Sn; and E = S, Se, Te) exhibit directional white light emission or produce second harmonics when irradiated with a continuous wave infrared laser source. The nature of the nonlinear optical properties correlates with the macroscopic structures of the cluster materials. The desired white light emission predominantly occurs in amorphous materials. It is therefore crucial to understand the geometric structures of the materials and the order within the materials. Here, we investigate the geometric structures of 12 different adamantane-like cluster materials by molecular dynamics simulations using a nonperiodic particle approach. The comparison of the calculated structure factors for two cluster materials with the corresponding experimental data obtained from diffraction and EXAFS measurements shows very good agreement. Our computations revealed that, on the one hand, larger, more flexible core structures (Ad < {Si4S6} < {Ge4S6} < {Sn4S6}) tend to lead to amorphous solids. On the other hand, larger substituents (methyl < phenyl < naphthyl) lead to more defined nearest neighbor interactions, with a tendency toward crystalline solids. Overall, our results show that a beginning order in the material results from a combination of the degree of flexibility of the core structure and the variation of the nearest neighbor interaction determined by the substituents.
中文翻译:

利用分子动力学模拟对类金刚烷簇材料非晶结构进行理论研究和比较
AdR 4 (Ad = 金刚烷,R = 有机取代基) 和 [(RT) 4 E 6 ] (R = 有机取代基;T = Si、Ge、Sn;且 E = S、Se、Te) 组成的簇材料表现出当用连续波红外激光源照射时,定向白光发射或产生二次谐波。非线性光学性质的本质与团簇材料的宏观结构相关。所需的白光发射主要发生在非晶材料中。因此,了解材料的几何结构和材料内部的顺序至关重要。在这里,我们使用非周期粒子方法通过分子动力学模拟研究了 12 种不同的金刚烷类簇材料的几何结构。将两种簇材料的计算结构因子与从衍射和 EXAFS 测量获得的相应实验数据进行比较,结果显示出非常好的一致性。我们的计算表明,一方面,更大、更灵活的核心结构(Ad < {Si 4 S 6 } < {Ge 4 S 6 } < {Sn 4 S 6 })往往会导致无定形固体。另一方面,较大的取代基(甲基%3C苯基%3C萘基)导致更明确的最近邻相互作用,并倾向于结晶固体。总的来说,我们的结果表明,材料的起始顺序是由核心结构的柔性程度和由取代基决定的最近邻相互作用的变化相结合的结果。
更新日期:2024-08-29
中文翻译:
利用分子动力学模拟对类金刚烷簇材料非晶结构进行理论研究和比较
AdR 4 (Ad = 金刚烷,R = 有机取代基) 和 [(RT) 4 E 6 ] (R = 有机取代基;T = Si、Ge、Sn;且 E = S、Se、Te) 组成的簇材料表现出当用连续波红外激光源照射时,定向白光发射或产生二次谐波。非线性光学性质的本质与团簇材料的宏观结构相关。所需的白光发射主要发生在非晶材料中。因此,了解材料的几何结构和材料内部的顺序至关重要。在这里,我们使用非周期粒子方法通过分子动力学模拟研究了 12 种不同的金刚烷类簇材料的几何结构。将两种簇材料的计算结构因子与从衍射和 EXAFS 测量获得的相应实验数据进行比较,结果显示出非常好的一致性。我们的计算表明,一方面,更大、更灵活的核心结构(Ad < {Si 4 S 6 } < {Ge 4 S 6 } < {Sn 4 S 6 })往往会导致无定形固体。另一方面,较大的取代基(甲基%3C苯基%3C萘基)导致更明确的最近邻相互作用,并倾向于结晶固体。总的来说,我们的结果表明,材料的起始顺序是由核心结构的柔性程度和由取代基决定的最近邻相互作用的变化相结合的结果。






























 京公网安备 11010802027423号
京公网安备 11010802027423号