当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Basis-Set Limit CCSD(T) Energies for Large Molecules with Local Natural Orbitals and Reduced-Scaling Basis-Set Corrections
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-29 , DOI: 10.1021/acs.jctc.4c00777 Dávid Mester 1, 2, 3 , Péter R Nagy 1, 2, 3 , Mihály Kállay 1, 2, 3
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-29 , DOI: 10.1021/acs.jctc.4c00777 Dávid Mester 1, 2, 3 , Péter R Nagy 1, 2, 3 , Mihály Kállay 1, 2, 3
Affiliation
|
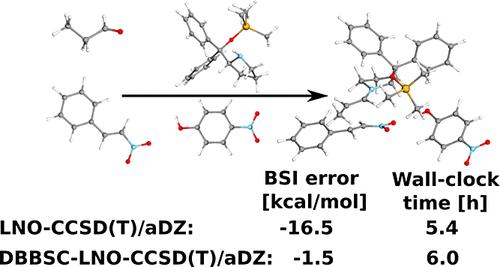
The calculation of density-based basis-set correction (DBBSC), which remedies the basis-set incompleteness (BSI) error of the correlation energy, is combined with local approximations. Aiming at large-scale applications, the procedure is implemented in our efficient local natural orbital-based coupled-cluster singles and doubles with perturbative triples [LNO-CCSD(T)] scheme. To this end, the range-separation function, which characterizes the one-electron BSI in space, is decomposed into the sum of contributions from individual localized molecular orbitals (LMOs). A compact domain is constructed around each LMO, and the corresponding contributions are evaluated only within these restricted domains. Furthermore, for the calculation of the complementary auxiliary basis set (CABS) correction, which significantly improves the Hartree–Fock (HF) energy, the local density fitting approximation is utilized. The errors arising from the local approximations are examined in detail, efficient prescreening techniques are introduced to compress the numerical quadrature used for DBBSC, and conservative default thresholds are selected for the truncation parameters. The efficiency of the DBBSC-LNO-CCSD(T) method is demonstrated through representative examples of up to 1000 atoms. Based on the numerical results, we conclude that the corrections drastically reduce the BSI error using double-ζ basis sets, often to below 1 kcal/mol compared to the reliable LNO-CCSD(T) complete basis set references, while significant improvements are also achieved with triple-ζ basis sets. Considering that the calculation of the DBBSC and CABS corrections only moderately increases the wall-clock time required for the post-HF steps in practical applications, the proposed DBBSC-LNO-CCSD(T) method offers a highly efficient and robust tool for large-scale calculations.
中文翻译:

具有局部自然轨道和缩小尺度的基组校正的大分子的基组极限 CCSD(T) 能量
基于密度的基组校正(DBBSC)的计算与局部近似相结合,可以纠正相关能量的基组不完整性(BSI)误差。针对大规模应用,该过程在我们高效的基于局部自然轨道的耦合团簇单团和双团与扰动三元组[LNO-CCSD(T)]方案中实现。为此,表征空间中单电子 BSI 的距离分离函数被分解为各个局域分子轨道 (LMO) 贡献的总和。围绕每个改性活生物体构建了一个紧凑的域,并且仅在这些受限域内评估相应的贡献。此外,为了计算互补辅助基组(CABS)校正,显着提高了 Hartree-Fock(HF)能量,利用了局部密度拟合近似。详细检查了局部近似产生的误差,引入了有效的预筛选技术来压缩用于 DBBSC 的数值求积,并为截断参数选择了保守的默认阈值。 DBBSC-LNO-CCSD(T) 方法的效率通过最多 1000 个原子的代表性示例得到证明。根据数值结果,我们得出的结论是,与可靠的 LNO-CCSD(T) 完整基组参考相比,使用双 ze 基组的修正大大降低了 BSI 误差,通常低于 1 kcal/mol,同时还显着改进了通过三重 z 基组实现。 考虑到实际应用中 DBBSC 和 CABS 修正的计算只会适度增加 HF 后步骤所需的挂钟时间,所提出的 DBBSC-LNO-CCSD(T) 方法为大规模计算提供了一种高效且稳健的工具。规模计算。
更新日期:2024-08-29
中文翻译:
具有局部自然轨道和缩小尺度的基组校正的大分子的基组极限 CCSD(T) 能量
基于密度的基组校正(DBBSC)的计算与局部近似相结合,可以纠正相关能量的基组不完整性(BSI)误差。针对大规模应用,该过程在我们高效的基于局部自然轨道的耦合团簇单团和双团与扰动三元组[LNO-CCSD(T)]方案中实现。为此,表征空间中单电子 BSI 的距离分离函数被分解为各个局域分子轨道 (LMO) 贡献的总和。围绕每个改性活生物体构建了一个紧凑的域,并且仅在这些受限域内评估相应的贡献。此外,为了计算互补辅助基组(CABS)校正,显着提高了 Hartree-Fock(HF)能量,利用了局部密度拟合近似。详细检查了局部近似产生的误差,引入了有效的预筛选技术来压缩用于 DBBSC 的数值求积,并为截断参数选择了保守的默认阈值。 DBBSC-LNO-CCSD(T) 方法的效率通过最多 1000 个原子的代表性示例得到证明。根据数值结果,我们得出的结论是,与可靠的 LNO-CCSD(T) 完整基组参考相比,使用双 ze 基组的修正大大降低了 BSI 误差,通常低于 1 kcal/mol,同时还显着改进了通过三重 z 基组实现。 考虑到实际应用中 DBBSC 和 CABS 修正的计算只会适度增加 HF 后步骤所需的挂钟时间,所提出的 DBBSC-LNO-CCSD(T) 方法为大规模计算提供了一种高效且稳健的工具。规模计算。






























 京公网安备 11010802027423号
京公网安备 11010802027423号