当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Multi-GPU RI-HF Energies and Analytic Gradients─Toward High-Throughput Ab Initio Molecular Dynamics
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-27 , DOI: 10.1021/acs.jctc.4c00877 Ryan Stocks 1 , Elise Palethorpe 1 , Giuseppe M J Barca 2, 3
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-27 , DOI: 10.1021/acs.jctc.4c00877 Ryan Stocks 1 , Elise Palethorpe 1 , Giuseppe M J Barca 2, 3
Affiliation
|
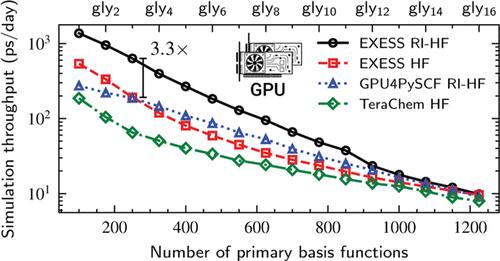
This article presents an optimized algorithm and implementation for calculating resolution-of-the-identity Hartree–Fock (RI-HF) energies and analytic gradients using multiple graphics processing units (GPUs). The algorithm is especially designed for high throughput ab initio molecular dynamics simulations of small and medium size molecules (10–100 atoms). Key innovations of this work include the exploitation of multi-GPU parallelism and a workload balancing scheme that efficiently distributes computational tasks among GPUs. Our implementation also employs techniques for symmetry utilization, integral screening, and leveraging sparsity to optimize memory usage. Computational results show that the implementation achieves significant performance improvements, including over 3 × speedups in single GPU AIMD throughput compared to previous GPU-accelerated RI-HF and traditional HF methods. Furthermore, utilizing multiple GPUs can provide superlinear speedup when the additional aggregate GPU memory allows for the storage of decompressed three-center integrals.
中文翻译:

多 GPU RI-HF 能量和分析梯度─迈向高通量从头算分子动力学
本文提出了一种优化算法和实现,用于使用多个图形处理单元 (GPU) 计算恒等分辨率 Hartree–Fock (RI-HF) 能量和分析梯度。该算法专为中小型分子(10-100 个原子)的高通量从头算分子动力学模拟而设计。这项工作的关键创新包括利用多 GPU 并行性以及在 GPU 之间有效分配计算任务的工作负载平衡方案。我们的实现还采用了对称利用、积分筛选和利用稀疏性来优化内存使用的技术。计算结果表明,该实现实现了显着的性能改进,与之前的 GPU 加速 RI-HF 和传统 HF 方法相比,单 GPU AIMD 吞吐量加速超过 3 倍。此外,当额外的聚合 GPU 内存允许存储解压的三中心积分时,利用多个 GPU 可以提供超线性加速。
更新日期:2024-08-27
中文翻译:
多 GPU RI-HF 能量和分析梯度─迈向高通量从头算分子动力学
本文提出了一种优化算法和实现,用于使用多个图形处理单元 (GPU) 计算恒等分辨率 Hartree–Fock (RI-HF) 能量和分析梯度。该算法专为中小型分子(10-100 个原子)的高通量从头算分子动力学模拟而设计。这项工作的关键创新包括利用多 GPU 并行性以及在 GPU 之间有效分配计算任务的工作负载平衡方案。我们的实现还采用了对称利用、积分筛选和利用稀疏性来优化内存使用的技术。计算结果表明,该实现实现了显着的性能改进,与之前的 GPU 加速 RI-HF 和传统 HF 方法相比,单 GPU AIMD 吞吐量加速超过 3 倍。此外,当额外的聚合 GPU 内存允许存储解压的三中心积分时,利用多个 GPU 可以提供超线性加速。






























 京公网安备 11010802027423号
京公网安备 11010802027423号