当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Crystal structure prediction and property calculation of copper–oxygen compounds using innovative search software from first principles
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-08-27 , DOI: 10.1039/d4cp02501f
Jinrong Huo 1, 2 , Kai Zhang 1, 2 , Pengfei Liu 1, 2 , Haocong Wei 1, 2 , Chaozheng He 2
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-08-27 , DOI: 10.1039/d4cp02501f
Jinrong Huo 1, 2 , Kai Zhang 1, 2 , Pengfei Liu 1, 2 , Haocong Wei 1, 2 , Chaozheng He 2
Affiliation
|
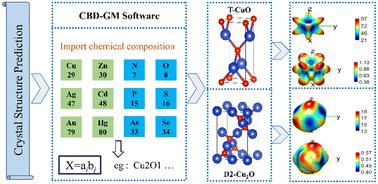
A Bayesian optimisation algorithm for deep learning crystal structure prediction software (CBD-GM) is used to predict the structures of Cu(I) and Cu(II) oxides of 2D and 3D materials. Two known 2D structures and two known 3D structures were anticipated, in addition to the prediction of 5 novel structures. All nine structures were optimised and analysed using density-functional theory (DFT). Firstly, DFT calculations using the PBE functional indicate that the structures should be thermodynamically and dynamically stable. Secondly, we calculated the elastic constants using the “stress–strain” method, and the predicted Young's modulus and Poisson's ratios of the materials suggest that they all should have excellent ductile mechanical properties. Calculations of the band structure of the materials performed using the Heyd–Scuseria–Ernzerhof (HSE) hybrid functional indicate that some of the materials should be semiconductors with useful bandgaps. The results therefore provide inspiration for the synthesis of new copper oxides for industrial applications.
中文翻译:

使用创新搜索软件从第一原理预测铜氧化合物的晶体结构和性质计算
深度学习晶体结构预测软件(CBD-GM)的贝叶斯优化算法用于预测2D和3D材料的Cu()和Cu( II )氧化物的结构。除了预测 5 个新颖结构外,还预计有两个已知的 2D 结构和两个已知的 3D 结构。所有九种结构均使用密度泛函理论(DFT)进行优化和分析。首先,使用 PBE 泛函的 DFT 计算表明结构应该是热力学和动态稳定的。其次,我们使用“应力-应变”方法计算了弹性常数,预测的材料的杨氏模量和泊松比表明它们都应具有优异的延性力学性能。使用 Heyd-Scuseria-Ernzerhof (HSE) 混合泛函对材料的能带结构进行的计算表明,某些材料应该是具有有用带隙的半导体。因此,这些结果为合成用于工业应用的新型铜氧化物提供了灵感。
更新日期:2024-08-27
中文翻译:
使用创新搜索软件从第一原理预测铜氧化合物的晶体结构和性质计算
深度学习晶体结构预测软件(CBD-GM)的贝叶斯优化算法用于预测2D和3D材料的Cu()和Cu( II )氧化物的结构。除了预测 5 个新颖结构外,还预计有两个已知的 2D 结构和两个已知的 3D 结构。所有九种结构均使用密度泛函理论(DFT)进行优化和分析。首先,使用 PBE 泛函的 DFT 计算表明结构应该是热力学和动态稳定的。其次,我们使用“应力-应变”方法计算了弹性常数,预测的材料的杨氏模量和泊松比表明它们都应具有优异的延性力学性能。使用 Heyd-Scuseria-Ernzerhof (HSE) 混合泛函对材料的能带结构进行的计算表明,某些材料应该是具有有用带隙的半导体。因此,这些结果为合成用于工业应用的新型铜氧化物提供了灵感。
































 京公网安备 11010802027423号
京公网安备 11010802027423号