当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hydrogen, Oxygen, and Lead Adsorbates on Al13Co4(100): Accurate Potential Energy Surfaces at Low Computational Cost by Machine Learning and DFT-Based Data
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-19 , DOI: 10.1021/acs.jctc.4c00367 Nathan Boulangeot 1, 2 , Florian Brix 1, 3 , Frédéric Sur 2 , Émilie Gaudry 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-19 , DOI: 10.1021/acs.jctc.4c00367 Nathan Boulangeot 1, 2 , Florian Brix 1, 3 , Frédéric Sur 2 , Émilie Gaudry 1
Affiliation
|
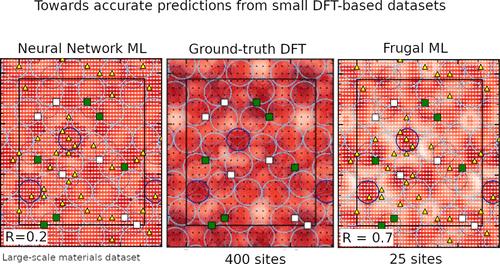
Intermetallic compounds are promising materials in numerous fields, especially those involving surface interactions, such as catalysis. A key factor to investigate their surface properties lies in adsorption energy maps, typically built using first-principles approaches. However, exploring the adsorption energy landscapes of intermetallic compounds can be cumbersome, usually requiring huge computational resources. In this work, we propose an efficient method to predict adsorption energies, based on a Machine Learning (ML) scheme fed by a few Density Functional Theory (DFT) estimates performed on n sites selected through the Farthest Point Sampling (FPS) process. We detail its application on the Al13Co4(100) quasicrystalline approximant surface for several atomic adsorbates (H, O, and Pb). On this specific example, our approach is shown to outperform both simple interpolation strategies and the recent ML force field MACE [arXiv.2206.07697], especially when the number n is small, i.e., below 36 sites. The ground-truth DFT adsorption energies are much more correlated with the predicted FPS-ML estimates (Pearson R-factor of 0.71, 0.73, and 0.90 for H, O and Pb, respectively, when n = 36) than with interpolation-based or MACE-ML ones (Pearson R-factors of 0.43, 0.39, and 0.56 for H, O, and Pb, in the former case and 0.22, 0.35, and 0.63 in the latter case). The unbiased root-mean-square error (ubRMSE) is lower for FPS-ML than for interpolation-based and MACE-ML predictions (0.15, 0.17, and 0.17 eV, respectively, for hydrogen and 0.17, 0.25, and 0.22 eV for lead), except for oxygen (0.55, 0.47, and 0.46 eV) due to large surface relaxations in this case. We believe that these findings and the corresponding methodology can be extended to a wide range of systems, which will motivate the discovery of novel functional materials.
中文翻译:

Al13Co4(100) 上的氢、氧和铅吸附物:通过机器学习和基于 DFT 的数据以低计算成本获得准确的势能面
金属间化合物是许多领域有前途的材料,特别是那些涉及表面相互作用的领域,例如催化。研究其表面特性的关键因素在于吸附能图,通常使用第一原理方法构建。然而,探索金属间化合物的吸附能景观可能很麻烦,通常需要大量的计算资源。在这项工作中,我们提出了一种预测吸附能的有效方法,该方法基于机器学习(ML)方案,该方案由对通过最远点采样(FPS)过程选择的n个站点进行的一些密度泛函理论(DFT)估计提供。我们详细介绍了其在几种原子吸附物(H、O 和 Pb)的 Al 13 Co 4 (100) 准晶近似表面上的应用。在这个具体的例子中,我们的方法被证明优于简单的插值策略和最近的 ML 力场 MACE [arXiv.2206.07697],特别是当数量n很小时,即低于 36 个站点时。与基于插值的或MACE-ML(前一种情况下 H、O 和 Pb 的 Pearson R 因子分别为 0.43、0.39 和 0.56,后一种情况下分别为 0.22、0.35 和 0.63)。 FPS-ML 的无偏均方根误差 (ubRMSE) 低于基于插值和 MACE-ML 的预测(氢分别为 0.15、0.17 和 0.17 eV,铅分别为 0.17、0.25 和 0.22 eV) ),但由于这种情况下表面弛豫较大,氧(0.55、0.47 和 0.46 eV)除外。 我们相信这些发现和相应的方法可以扩展到广泛的系统,这将推动新型功能材料的发现。
更新日期:2024-08-19
中文翻译:
Al13Co4(100) 上的氢、氧和铅吸附物:通过机器学习和基于 DFT 的数据以低计算成本获得准确的势能面
金属间化合物是许多领域有前途的材料,特别是那些涉及表面相互作用的领域,例如催化。研究其表面特性的关键因素在于吸附能图,通常使用第一原理方法构建。然而,探索金属间化合物的吸附能景观可能很麻烦,通常需要大量的计算资源。在这项工作中,我们提出了一种预测吸附能的有效方法,该方法基于机器学习(ML)方案,该方案由对通过最远点采样(FPS)过程选择的n个站点进行的一些密度泛函理论(DFT)估计提供。我们详细介绍了其在几种原子吸附物(H、O 和 Pb)的 Al 13 Co 4 (100) 准晶近似表面上的应用。在这个具体的例子中,我们的方法被证明优于简单的插值策略和最近的 ML 力场 MACE [arXiv.2206.07697],特别是当数量n很小时,即低于 36 个站点时。与基于插值的或MACE-ML(前一种情况下 H、O 和 Pb 的 Pearson R 因子分别为 0.43、0.39 和 0.56,后一种情况下分别为 0.22、0.35 和 0.63)。 FPS-ML 的无偏均方根误差 (ubRMSE) 低于基于插值和 MACE-ML 的预测(氢分别为 0.15、0.17 和 0.17 eV,铅分别为 0.17、0.25 和 0.22 eV) ),但由于这种情况下表面弛豫较大,氧(0.55、0.47 和 0.46 eV)除外。 我们相信这些发现和相应的方法可以扩展到广泛的系统,这将推动新型功能材料的发现。
































 京公网安备 11010802027423号
京公网安备 11010802027423号