当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Methods for Classical-Mechanical Molecular Simulation in Chemistry: Achievements, Limitations, Perspectives
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-08-13 , DOI: 10.1021/acs.jcim.4c00823 Wilfred F van Gunsteren 1 , Chris Oostenbrink 2, 3
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-08-13 , DOI: 10.1021/acs.jcim.4c00823 Wilfred F van Gunsteren 1 , Chris Oostenbrink 2, 3
Affiliation
|
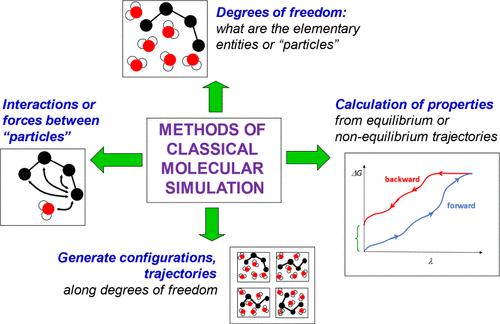
More than a half century ago it became feasible to simulate, using classical-mechanical equations of motion, the dynamics of molecular systems on a computer. Since then classical-physical molecular simulation has become an integral part of chemical research. It is widely applied in a variety of branches of chemistry and has significantly contributed to the development of chemical knowledge. It offers understanding and interpretation of experimental results, semiquantitative predictions for measurable and nonmeasurable properties of substances, and allows the calculation of properties of molecular systems under conditions that are experimentally inaccessible. Yet, molecular simulation is built on a number of assumptions, approximations, and simplifications which limit its range of applicability and its accuracy. These concern the potential-energy function used, adequate sampling of the vast statistical-mechanical configurational space of a molecular system and the methods used to compute particular properties of chemical systems from statistical-mechanical ensembles. During the past half century various methodological ideas to improve the efficiency and accuracy of classical-physical molecular simulation have been proposed, investigated, evaluated, implemented in general simulation software or were abandoned. The latter because of fundamental flaws or, while being physically sound, computational inefficiency. Some of these methodological ideas are briefly reviewed and the most effective methods are highlighted. Limitations of classical-physical simulation are discussed and perspectives are sketched.
中文翻译:

化学中的经典力学分子模拟方法:成就、局限性、前景
半个多世纪前,使用经典力学运动方程在计算机上模拟分子系统的动力学变得可行。从那时起,经典物理分子模拟已成为化学研究不可或缺的一部分。它广泛应用于化学的各个分支,对化学知识的发展做出了重大贡献。它提供对实验结果的理解和解释,对物质的可测量和不可测量特性的半定量预测,并允许在实验无法达到的条件下计算分子系统的特性。然而,分子模拟建立在许多假设、近似和简化的基础上,这限制了其适用范围和准确性。这些涉及所使用的势能函数、分子系统巨大统计机械构型空间的充分采样以及用于从统计机械系综计算化学系统特定性质的方法。在过去的半个世纪中,各种提高经典物理分子模拟效率和准确性的方法论思想已经被提出、研究、评估、在通用模拟软件中实施或被放弃。后者是因为根本性缺陷,或者虽然物理上合理,但计算效率低下。简要回顾了其中一些方法论思想,并强调了最有效的方法。讨论了经典物理模拟的局限性并概述了前景。
更新日期:2024-08-13
中文翻译:
化学中的经典力学分子模拟方法:成就、局限性、前景
半个多世纪前,使用经典力学运动方程在计算机上模拟分子系统的动力学变得可行。从那时起,经典物理分子模拟已成为化学研究不可或缺的一部分。它广泛应用于化学的各个分支,对化学知识的发展做出了重大贡献。它提供对实验结果的理解和解释,对物质的可测量和不可测量特性的半定量预测,并允许在实验无法达到的条件下计算分子系统的特性。然而,分子模拟建立在许多假设、近似和简化的基础上,这限制了其适用范围和准确性。这些涉及所使用的势能函数、分子系统巨大统计机械构型空间的充分采样以及用于从统计机械系综计算化学系统特定性质的方法。在过去的半个世纪中,各种提高经典物理分子模拟效率和准确性的方法论思想已经被提出、研究、评估、在通用模拟软件中实施或被放弃。后者是因为根本性缺陷,或者虽然物理上合理,但计算效率低下。简要回顾了其中一些方法论思想,并强调了最有效的方法。讨论了经典物理模拟的局限性并概述了前景。






























 京公网安备 11010802027423号
京公网安备 11010802027423号