当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
SPRank─A Knowledge-Based Scoring Function for RNA-Ligand Pose Prediction and Virtual Screening
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-16 , DOI: 10.1021/acs.jctc.4c00681 Yuanzhe Zhou 1 , Yangwei Jiang 1 , Shi-Jie Chen 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-08-16 , DOI: 10.1021/acs.jctc.4c00681 Yuanzhe Zhou 1 , Yangwei Jiang 1 , Shi-Jie Chen 2
Affiliation
|
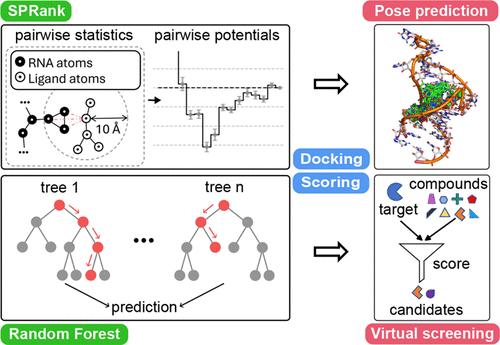
The growing interest in RNA-targeted drugs underscores the need for computational modeling of interactions between RNA molecules and small compounds. Having a reliable scoring function for RNA-ligand interactions is essential for effective computational drug screening. An ideal scoring function should not only predict the native pose for ligand binding but also rank the affinity of the binding for different ligands. However, existing scoring functions are primarily designed to predict the native binding modes for a given RNA-ligand pair and have not been thoroughly assessed for virtual screening purposes. In this paper, we introduce SPRank, a combination of machine-learning and knowledge-based scoring functions developed through a weighted iterative approach, specifically designed to tackle both binding mode prediction and virtual screening challenges. Our approach incorporates third-party docking software, such as rDock and AutoDock Vina, to sample flexible ligands against an ensemble of RNA structures, capturing the conformational flexibility of both the RNA and the ligand. Through rigorous testing, SPRank demonstrates improved performance compared to the tested scoring functions across four test sets comprising 122, 42, 55, and 71 nucleic acid-ligand complexes. Furthermore, SPRank exhibits improved performance in virtual screening tests targeting the HIV-1 TAR ensemble, which highlights its advantage in drug discovery. These results underscore the advantages of SPRank as a potentially promising tool for the RNA-targeted drug design. The source code of SPRank and the data sets are freely accessible at https://github.com/Vfold-RNA/SPRank.
中文翻译:

SPRank─用于 RNA 配体姿势预测和虚拟筛选的基于知识的评分函数
人们对 RNA 靶向药物日益增长的兴趣强调了对 RNA 分子和小化合物之间相互作用进行计算建模的必要性。拥有可靠的 RNA-配体相互作用评分函数对于有效的计算药物筛选至关重要。理想的评分函数不仅应该预测配体结合的天然姿势,还应该对不同配体的结合亲和力进行排序。然而,现有的评分函数主要设计用于预测给定 RNA-配体对的天然结合模式,并且尚未针对虚拟筛选目的进行彻底评估。在本文中,我们介绍了 SPRank,它是通过加权迭代方法开发的机器学习和基于知识的评分函数的组合,专门用于解决结合模式预测和虚拟筛选挑战。我们的方法结合了第三方对接软件,例如 rDock 和 AutoDock Vina,针对一组 RNA 结构对灵活的配体进行采样,捕获 RNA 和配体的构象灵活性。通过严格的测试,与包含 122、42、55 和 71 个核酸-配体复合物的四个测试集的测试评分函数相比,SPRank 表现出更高的性能。此外,SPRank 在针对 HIV-1 TAR 整体的虚拟筛选测试中表现出改进的性能,这凸显了其在药物发现方面的优势。这些结果强调了 SPRank 作为 RNA 靶向药物设计的潜在有前途工具的优势。 SPRank 的源代码和数据集可在 https://github.com/Vfold-RNA/SPRank 上免费获取。
更新日期:2024-08-16
中文翻译:
SPRank─用于 RNA 配体姿势预测和虚拟筛选的基于知识的评分函数
人们对 RNA 靶向药物日益增长的兴趣强调了对 RNA 分子和小化合物之间相互作用进行计算建模的必要性。拥有可靠的 RNA-配体相互作用评分函数对于有效的计算药物筛选至关重要。理想的评分函数不仅应该预测配体结合的天然姿势,还应该对不同配体的结合亲和力进行排序。然而,现有的评分函数主要设计用于预测给定 RNA-配体对的天然结合模式,并且尚未针对虚拟筛选目的进行彻底评估。在本文中,我们介绍了 SPRank,它是通过加权迭代方法开发的机器学习和基于知识的评分函数的组合,专门用于解决结合模式预测和虚拟筛选挑战。我们的方法结合了第三方对接软件,例如 rDock 和 AutoDock Vina,针对一组 RNA 结构对灵活的配体进行采样,捕获 RNA 和配体的构象灵活性。通过严格的测试,与包含 122、42、55 和 71 个核酸-配体复合物的四个测试集的测试评分函数相比,SPRank 表现出更高的性能。此外,SPRank 在针对 HIV-1 TAR 整体的虚拟筛选测试中表现出改进的性能,这凸显了其在药物发现方面的优势。这些结果强调了 SPRank 作为 RNA 靶向药物设计的潜在有前途工具的优势。 SPRank 的源代码和数据集可在 https://github.com/Vfold-RNA/SPRank 上免费获取。






























 京公网安备 11010802027423号
京公网安备 11010802027423号