当前位置:
X-MOL 学术
›
Catal. Sci. Technol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
First principles investigation of manganese catalyst structure and coordination in the p-xylene oxidation process
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2024-08-15 , DOI: 10.1039/d4cy00284a Harry N. Thomas 1 , Duncan F. Wass 1 , Caroline A. Offiler 2 , Keith Whiston 2 , Andrew J. Logsdail 1
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2024-08-15 , DOI: 10.1039/d4cy00284a Harry N. Thomas 1 , Duncan F. Wass 1 , Caroline A. Offiler 2 , Keith Whiston 2 , Andrew J. Logsdail 1
Affiliation
|
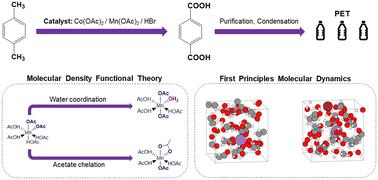
The oxidation of p-xylene to terephtalic acid has global importance, with the product used as a precursor for polyethylene terephthalate (PET). The oxidation of p-xylene proceeds via a redox cascade that involves cobalt, manganese, and bromide, with a synergy allowing for high selectivity and reactivity; however, the equilibrium coordination environment of the catalyst species remains uncertain due to the hostile industrial operating conditions. To build knowledge of the catalyst speciation and develop understanding of the reaction process, a density functional theory approach is applied herein to determine the static and dynamic properties of the divalent (reduced) and trivalent (oxidized) manganese catalysts in the redox cascade. The Gibbs free energy has been calculated for manganese as a function of ligands in the inner coordination sphere, with the octahedrally-coordinated Mn(OAc)2(HOAc)2 and Mn(OAc)3(H2O)1 identified as the most thermodynamically stable coordination environments for Mn(II) and Mn(III), respectively. Dynamic properties of these catalysts in the presence of an explicit solvent environment have been determined using first principles molecular dynamics simulations. The simulations indicate 0–2 coordinating water ligands are present in the inner coordination sphere under standard industrial temperatures and pressures. The dynamical simulations have been extended to include HBr, which couples with Mn in the redox cascade, and the bromide species does not enter in the inner-coordination sphere of the oxidized Mn(III) catalyst, providing evidence that the electron transfer between bromide and Mn(III) proceeds via an outer sphere mechanism. Our results suggest that oxidation of Mn(II) has the potential for facilitating L-type ligand exchange in the inner-sphere coordination environment. The results are a platform for developing a more complete knowledge of the reaction mechanism at the atomistic scale.
中文翻译:

对二甲苯氧化过程中锰催化剂结构和配位的第一性原理研究
对二甲苯氧化成对苯二甲酸具有全球重要性,该产品用作聚对苯二甲酸乙二醇酯 (PET) 的前体。对二甲苯的氧化通过涉及钴、锰和溴化物的氧化还原级联进行,具有协同作用,可实现高选择性和反应性;然而,由于恶劣的工业操作条件,催化剂物种的平衡配位环境仍然不确定。为了建立对催化剂形态的了解并加深对反应过程的理解,本文应用密度泛函理论方法来确定氧化还原级联中二价(还原)和三价(氧化)锰催化剂的静态和动态性质。锰的吉布斯自由能作为内配位层配体的函数进行了计算,其中八面体配位的 Mn(OAc) 2 (HOAc) 2和 Mn(OAc) 3 (H 2 O) 1被认为是最有效的。分别为 Mn( II ) 和 Mn( III ) 的热力学稳定配位环境。使用第一原理分子动力学模拟确定了这些催化剂在明确溶剂环境下的动态特性。模拟表明,在标准工业温度和压力下,内部配位层中存在 0-2 个配位水配体。 动力学模拟已扩展到包括 HBr,它在氧化还原级联中与 Mn 偶联,并且溴化物物质不会进入氧化 Mn( III ) 催化剂的内配位层,这提供了溴化物与 Mn(III) 催化剂之间的电子转移的证据。 Mn( III )通过外球机制进行。我们的结果表明,Mn( II ) 的氧化具有促进内球配位环境中 L 型配体交换的潜力。这些结果为在原子尺度上开发更完整的反应机理知识提供了一个平台。
更新日期:2024-08-15
中文翻译:
对二甲苯氧化过程中锰催化剂结构和配位的第一性原理研究
对二甲苯氧化成对苯二甲酸具有全球重要性,该产品用作聚对苯二甲酸乙二醇酯 (PET) 的前体。对二甲苯的氧化通过涉及钴、锰和溴化物的氧化还原级联进行,具有协同作用,可实现高选择性和反应性;然而,由于恶劣的工业操作条件,催化剂物种的平衡配位环境仍然不确定。为了建立对催化剂形态的了解并加深对反应过程的理解,本文应用密度泛函理论方法来确定氧化还原级联中二价(还原)和三价(氧化)锰催化剂的静态和动态性质。锰的吉布斯自由能作为内配位层配体的函数进行了计算,其中八面体配位的 Mn(OAc) 2 (HOAc) 2和 Mn(OAc) 3 (H 2 O) 1被认为是最有效的。分别为 Mn( II ) 和 Mn( III ) 的热力学稳定配位环境。使用第一原理分子动力学模拟确定了这些催化剂在明确溶剂环境下的动态特性。模拟表明,在标准工业温度和压力下,内部配位层中存在 0-2 个配位水配体。 动力学模拟已扩展到包括 HBr,它在氧化还原级联中与 Mn 偶联,并且溴化物物质不会进入氧化 Mn( III ) 催化剂的内配位层,这提供了溴化物与 Mn(III) 催化剂之间的电子转移的证据。 Mn( III )通过外球机制进行。我们的结果表明,Mn( II ) 的氧化具有促进内球配位环境中 L 型配体交换的潜力。这些结果为在原子尺度上开发更完整的反应机理知识提供了一个平台。




































 京公网安备 11010802027423号
京公网安备 11010802027423号