当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Photophysics in Biomembranes: Computational Insight into the Interaction between Lipid Bilayers and Chromophores
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2024-08-06 , DOI: 10.1021/acs.accounts.4c00153 S Osella 1 , S Knippenberg 2
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2024-08-06 , DOI: 10.1021/acs.accounts.4c00153 S Osella 1 , S Knippenberg 2
Affiliation
|
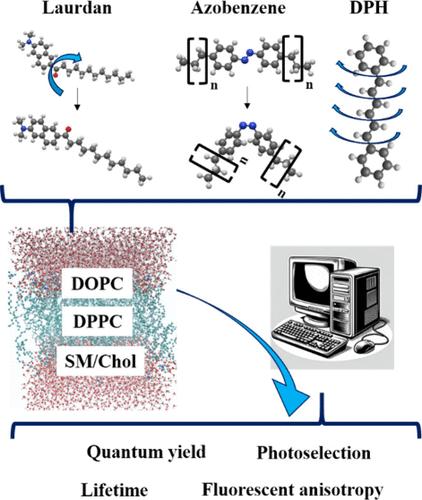
Light is ubiquitously available to probe the structure and dynamics of biomolecules and biological tissues. Generally, this cannot be done directly with visible light, because of the absence of absorption by those biomolecules. This problem can be overcome by incorporating organic molecules (chromophores) that show an optical response in the vicinity of those biomolecules. Since those optical properties are strongly dependent on the chromophore’s environment, time-resolved spectroscopic studies can provide a wealth of information on biosystems at the molecular scale in a nondestructive way. In this work, we give an overview on the multiscale computational strategy developed by us in the last eight years and prove that theoretical studies and simulations are needed to explain, guide, and predict observations in fluorescence experiments. As we challenge the accepted views on existing probes, we discover unexplored abilities that can discriminate surrounding lipid bilayers and their temperature-dependent as well as solvent-dependent properties. We focus on three archetypal chromophores: diphenylhexatriene (DPH), Laurdan, and azobenzene. Our method shows that conformational changes should not be neglected for the prototype rod-shaped molecule DPH. They determine its position and orientation in a liquid-ordered (Lo) sphingomyelin/cholesterol (SM/Chol) bilayer and are responsible for a strong differentiation of its absorption spectra and fluorescence decay times in dioleoylphosphatidylcholine (DOPC) and dipalmitoylphosphatidylcholine (DPPC) membranes, which are at room temperature in liquid-disordered (Ld) and solid-gel (So) phases, respectively. Thanks to its pronounced first excited state dipole moment, Laurdan has long been known as a solvatochromic probe. Since this molecule has however two conformers, we prove that they exhibit different properties in different lipid membrane phases. We see that the two conformers are only blocked in one phase but not in another. Supported by fluorescence anisotropy decay simulations, Laurdan can therefore be regarded as a molecular rotor. Finally, the conformational versatility of azobenzene in saturated Ld lipid bilayers is simulated, along with its photoisomerization pathways. By means of nonadiabatic QM/MM surface hopping analyses (QM/MM-SH), a dual mechanism is found with a torsional mechanism and a slow conversion for trans-to-cis. For cis-to-trans, simulations show a much higher quantum yield and a so-called “pedal-like” mechanism. The differences are related to the different potential energy surfaces as well as the interactions with the surrounding alkyl chains. When tails of increased length are attached to this probe, cis is pushed toward the polar surface, while trans is pulled toward the center of the membrane.
中文翻译:

生物膜中的光物理学:脂质双层和发色团之间相互作用的计算洞察
光无处不在,可用于探测生物分子和生物组织的结构和动力学。通常,这不能直接用可见光完成,因为这些生物分子没有吸收。这个问题可以通过掺入有机分子(发色团)来克服,这些有机分子在这些生物分子附近显示出光学响应。由于这些光学特性在很大程度上取决于发色团的环境,因此时间分辨光谱研究可以以非破坏性方式在分子尺度上提供有关生物系统的大量信息。在这项工作中,我们概述了我们在过去八年中开发的多尺度计算策略,并证明需要理论研究和模拟来解释、指导和预测荧光实验中的观察结果。当我们挑战现有探针的公认观点时,我们发现了尚未开发的能力,这些能力可以区分周围的脂质双层及其温度依赖性和溶剂依赖性特性。我们专注于三种原型发色团:二苯基己三烯 (DPH)、Laurdan 和偶氮苯。我们的方法表明,原型杆状分子 DPH 的构象变化不应被忽视。它们确定其在液体有序 (Lo) 鞘磷脂/胆固醇 (SM/Chol) 双层中的位置和方向,并负责其在二油酰磷脂酰胆碱 (DOPC) 和二棕榈酰磷脂酰胆碱 (DPPC) 膜中的吸收光谱和荧光衰减时间的强烈区分,它们在室温下分别处于液体无序 (Ld) 和固体凝胶 (So) 相。由于其明显的第一激发态偶极矩,Laurdan 长期以来一直被称为溶剂变色探针。 然而,由于该分子有两个构象异构体,我们证明它们在不同的脂质膜相中表现出不同的性质。我们看到这两个 conformer 仅在一个阶段被阻塞,而在另一个阶段中没有被阻塞。因此,在荧光各向异性衰变模拟的支持下,Laurdan 可以被视为分子转子。最后,模拟了偶氮苯在饱和 Ld 脂质双层中的构象多功能性,以及其光异构化途径。通过非绝热 QM/MM 表面跳跃分析 (QM/MM-SH),发现了具有扭转机制和反式到顺式缓慢转换的双重机制。对于 cis-to-trans,模拟显示更高的量子产率和所谓的“踏板状”机制。这些差异与不同的势能表面以及与周围烷基链的相互作用有关。当长度增加的尾部连接到该探针上时,顺式被推向极表面,而反式被拉向膜中心。
更新日期:2024-08-06
中文翻译:
生物膜中的光物理学:脂质双层和发色团之间相互作用的计算洞察
光无处不在,可用于探测生物分子和生物组织的结构和动力学。通常,这不能直接用可见光完成,因为这些生物分子没有吸收。这个问题可以通过掺入有机分子(发色团)来克服,这些有机分子在这些生物分子附近显示出光学响应。由于这些光学特性在很大程度上取决于发色团的环境,因此时间分辨光谱研究可以以非破坏性方式在分子尺度上提供有关生物系统的大量信息。在这项工作中,我们概述了我们在过去八年中开发的多尺度计算策略,并证明需要理论研究和模拟来解释、指导和预测荧光实验中的观察结果。当我们挑战现有探针的公认观点时,我们发现了尚未开发的能力,这些能力可以区分周围的脂质双层及其温度依赖性和溶剂依赖性特性。我们专注于三种原型发色团:二苯基己三烯 (DPH)、Laurdan 和偶氮苯。我们的方法表明,原型杆状分子 DPH 的构象变化不应被忽视。它们确定其在液体有序 (Lo) 鞘磷脂/胆固醇 (SM/Chol) 双层中的位置和方向,并负责其在二油酰磷脂酰胆碱 (DOPC) 和二棕榈酰磷脂酰胆碱 (DPPC) 膜中的吸收光谱和荧光衰减时间的强烈区分,它们在室温下分别处于液体无序 (Ld) 和固体凝胶 (So) 相。由于其明显的第一激发态偶极矩,Laurdan 长期以来一直被称为溶剂变色探针。 然而,由于该分子有两个构象异构体,我们证明它们在不同的脂质膜相中表现出不同的性质。我们看到这两个 conformer 仅在一个阶段被阻塞,而在另一个阶段中没有被阻塞。因此,在荧光各向异性衰变模拟的支持下,Laurdan 可以被视为分子转子。最后,模拟了偶氮苯在饱和 Ld 脂质双层中的构象多功能性,以及其光异构化途径。通过非绝热 QM/MM 表面跳跃分析 (QM/MM-SH),发现了具有扭转机制和反式到顺式缓慢转换的双重机制。对于 cis-to-trans,模拟显示更高的量子产率和所谓的“踏板状”机制。这些差异与不同的势能表面以及与周围烷基链的相互作用有关。当长度增加的尾部连接到该探针上时,顺式被推向极表面,而反式被拉向膜中心。






























 京公网安备 11010802027423号
京公网安备 11010802027423号