Cell Death and Differentiation ( IF 13.7 ) Pub Date : 2024-08-05 , DOI: 10.1038/s41418-024-01351-0 Xiang Luo 1 , Hai-Biao Gong 1 , Zi-Chun Li 1 , Dong-Dong Li 1 , Zi-Xuan Li 1 , Jie Sun 1 , Chang-Yu Yan 1 , Rui-Ting Huang 2 , Yue Feng 3 , Shu-Rui Chen 1 , Yun-Feng Cao 4 , Mingxian Liu 3 , Rong Wang 1, 5 , Feng Huang 5 , Wan-Yang Sun 1 , Hiroshi Kurihara 1 , Wen-Jun Duan 1 , Lei Liang 1 , Wen Jin 1 , Yan-Ping Wu 1 , Rong-Rong He 1, 2, 5 , Yi-Fang Li 1
|
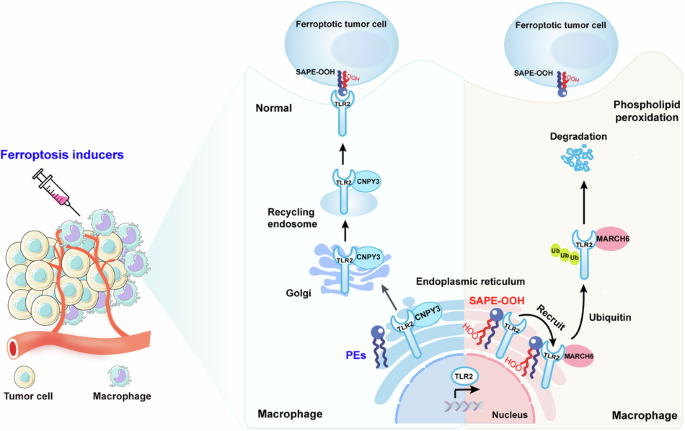
Ferroptosis holds significant potential for application in cancer therapy. However, ferroptosis inducers are not cell-specific and can cause phospholipid peroxidation in both tumor and non-tumor cells. This limitation greatly restricts the use of ferroptosis therapy as a safe and effective anticancer strategy. Our previous study demonstrated that macrophages can engulf ferroptotic cells through Toll-like receptor 2 (TLR2). Despite this advancement, the precise mechanism by which phospholipid peroxidation in macrophages affects their phagocytotic capability during treatment of tumors with ferroptotic agents is still unknown. Here, we utilized flow sorting combined with redox phospholipidomics to determine that phospholipid peroxidation in tumor microenvironment (TME) macrophages impaired the macrophages ability to eliminate ferroptotic tumor cells by phagocytosis, ultimately fostering tumor resistance to ferroptosis therapy. Mechanistically, the accumulation of phospholipid peroxidation in the macrophage endoplasmic reticulum (ER) repressed TLR2 trafficking to the plasma membrane and caused its retention in the ER by disrupting the interaction between TLR2 and its chaperone CNPY3. Subsequently, this ER-retained TLR2 recruited E3 ligase MARCH6 and initiated the proteasome-dependent degradation. Using redox phospholipidomics, we identified 1-steaoryl-2-15-HpETE-sn-glycero-3-phosphatidylethanolamine (SAPE-OOH) as the crucial mediator of these effects. Conclusively, our discovery elucidates a novel molecular mechanism underlying macrophage phospholipid peroxidation-induced tumor resistance to ferroptosis therapy and highlights the TLR2-MARCH6 axis as a potential therapeutic target for cancer therapy.
中文翻译:
巨噬细胞中的磷脂过氧化通过抑制对铁死亡细胞的吞噬能力来赋予肿瘤抗性
铁死亡在癌症治疗中具有巨大的应用潜力。然而,铁死亡诱导剂不是细胞特异性的,并且可以在肿瘤和非肿瘤细胞中引起磷脂过氧化。这一限制极大地限制了铁死亡疗法作为安全有效的抗癌策略的使用。我们之前的研究表明,巨噬细胞可以通过 Toll 样受体 2 (TLR2) 吞噬铁死亡细胞。尽管取得了这一进展,但在用铁死亡剂治疗肿瘤期间,巨噬细胞中的磷脂过氧化影响其吞噬能力的确切机制仍然未知。在这里,我们利用流式分选结合氧化还原磷脂组学来确定肿瘤微环境(TME)巨噬细胞中的磷脂过氧化损害了巨噬细胞通过吞噬作用消除铁死亡肿瘤细胞的能力,最终促进了肿瘤对铁死亡治疗的抵抗。从机制上讲,巨噬细胞内质网 (ER) 中磷脂过氧化的积累抑制了 TLR2 向质膜的运输,并通过破坏 TLR2 与其伴侣 CNPY3 之间的相互作用而导致其保留在 ER 中。随后,该 ER 保留的 TLR2 招募 E3 连接酶 MARCH6 并启动蛋白酶体依赖性降解。使用氧化还原磷脂组学,我们确定 1-硬脂酰基-2-15-HpETE-sn-甘油-3-磷脂酰乙醇胺 (SAPE-OOH) 是这些效应的关键介质。总之,我们的发现阐明了巨噬细胞磷脂过氧化诱导的肿瘤对铁死亡治疗产生抗性的新分子机制,并强调 TLR2-MARCH6 轴作为癌症治疗的潜在治疗靶点。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号