当前位置:
X-MOL 学术
›
J. Adv. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
NAT10 promotes osteoclastogenesis in inflammatory bone loss by catalyzing Fos mRNA ac4C modification and upregulating MAPK signaling pathway
Journal of Advanced Research ( IF 11.4 ) Pub Date : 2024-07-31 , DOI: 10.1016/j.jare.2024.07.031 Ruhan Yang 1 , Weijun Yu 1 , Lu Lin 1 , Zhurong Cui 1 , Jiaqi Tang 1 , Guanglong Li 1 , Min Jin 1 , Yuting Gu 1 , Eryi Lu 1
中文翻译:

NAT10 通过催化 Fos mRNA ac4C 修饰和上调 MAPK 信号通路促进炎性骨质流失中的破骨细胞生成
过度破骨细胞生成是炎性骨质流失的关键驱动因素。抑制破骨细胞生成一直被认为是治疗炎症性骨质流失所必需的。N-乙酰转移酶 10 (NAT10) 是唯一负责 mRNA 的 N4-乙酰胞苷 (ac4C) 修饰的酶,并参与细胞发育。然而,它在破骨细胞生成和炎性骨质流失中的作用仍然难以捉摸。
我们旨在阐明 NAT10 和 ac4C 修饰在破骨细胞生成和炎性骨质流失中的调控机制。
采用实时定量PCR(quantitative real-time PCR,qPCR)、western blotting、dot blot和免疫荧光染色法测定破骨细胞生成过程中NAT10的表达和ac4C修饰,采用抗酒石酸盐酸性磷酸酶染色、足小体带染色和骨吸收坑法测定NAT10抑制对破骨细胞分化的影响。然后,进行 acRIP-qPCR 和 NAT10RIP-qPCR、ac4C 位点预测、mRNA 衰变检测和荧光素酶报告基因分析,进一步研究其潜在机制。最后,应用炎症性骨质流失小鼠模型验证 NAT10 抑制在体内的治疗效果。
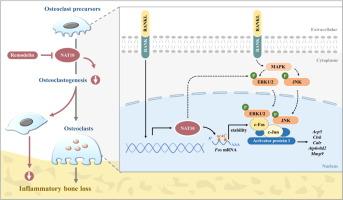
NAT10 表达在破骨细胞分化过程中上调,在牙周炎小鼠的牙槽骨破骨细胞中高表达。抑制 NAT10 显着降低了体外破骨细胞的分化,表现为酒石酸盐耐药性酸性磷酸酶阳性多核细胞、破骨细胞特异性基因表达、F-肌动蛋白环形成和骨吸收能力的大幅减少。从机制上讲,NAT10 催化了 Fos (编码 AP-1 组分 c-Fos) mRNA 的 ac4C 修饰并保持其稳定性。此外,NAT10 促进 MAPK 信号通路,从而激活 AP-1 (c-Fos/c-Jun) 转录进行破骨细胞生成。在治疗上,NAT10 的特异性抑制剂 Remodelin 的给药显着阻碍了结扎诱导的牙槽骨流失和脂多糖诱导的炎性颅骨溶解。
我们的研究表明,NAT10 介导的 ac4C 修饰是破骨细胞分化的重要表观遗传调控,并提出了一个有前途的炎症性骨质流失治疗靶点。
更新日期:2024-07-31
Journal of Advanced Research ( IF 11.4 ) Pub Date : 2024-07-31 , DOI: 10.1016/j.jare.2024.07.031 Ruhan Yang 1 , Weijun Yu 1 , Lu Lin 1 , Zhurong Cui 1 , Jiaqi Tang 1 , Guanglong Li 1 , Min Jin 1 , Yuting Gu 1 , Eryi Lu 1
Affiliation
|
Introduction
Excessive osteoclastogenesis is a key driver of inflammatory bone loss. Suppressing osteoclastogenesis has always been considered essential for the treatment of inflammatory bone loss. N-acetyltransferase 10 (NAT10) is the sole enzyme responsible for N4-acetylcytidine (ac4C) modification of mRNA, and is involved in cell development. However, its role in osteoclastogenesis and inflammatory bone loss remained elusive.Objectives
We aimed to clarify the regulatory mechanism of NAT10 and ac4C modification in osteoclastogenesis and inflammatory bone loss.Methods
NAT10 expression and ac4C modification during osteoclastogenesis were determined by quantitative real-time PCR (qPCR), western blotting, dot blot and immunofluorescent staining, and the effect of NAT10 inhibition on osteoclast differentiation in vitro was measured by the tartrate-resistant acid phosphatase staining, podosome belts staining assay and bone resorption pit assay. Then, acRIP-qPCR and NAT10RIP-qPCR, ac4C site prediction, mRNA decay assay and luciferase reporter assay were performed to further study the underlying mechanisms. At last, mice models of inflammatory bone loss were applied to verify the therapeutic effect of NAT10 inhibition in vivo.Results
NAT10 expression was upregulated during osteoclast differentiation and highly expressed in alveolar bone osteoclasts from periodontitis mice. Inhibition of NAT10 notably reduced osteoclast differentiation in vitro, as indicated by great reduction of tartrated resistant acid phosphatse positive multinuclear cells, osteoclast-specific gene expression, F-actin ring formation and bone resorption capacity. Mechanistically, NAT10 catalyzed ac4C modification of Fos (encoding AP-1 component c-Fos) mRNA and maintained its stabilization. Besides, NAT10 promoted MAPK signaling pathway and thereby activated AP-1 (c-Fos/c-Jun) transcription for osteoclastogenesis. Therapeutically, administration of Remodelin, the specific inhibitor of NAT10, remarkably impeded the ligature-induced alveolar bone loss and lipopolysaccharide-induced inflammatory calvarial osteolysis.Conclusions
Our study demonstrated that NAT10-mediated ac4C modification is an important epigenetic regulation of osteoclast differentiation and proposed a promising therapeutic target for inflammatory bone loss.中文翻译:
NAT10 通过催化 Fos mRNA ac4C 修饰和上调 MAPK 信号通路促进炎性骨质流失中的破骨细胞生成
介绍
过度破骨细胞生成是炎性骨质流失的关键驱动因素。抑制破骨细胞生成一直被认为是治疗炎症性骨质流失所必需的。N-乙酰转移酶 10 (NAT10) 是唯一负责 mRNA 的 N4-乙酰胞苷 (ac4C) 修饰的酶,并参与细胞发育。然而,它在破骨细胞生成和炎性骨质流失中的作用仍然难以捉摸。
目标
我们旨在阐明 NAT10 和 ac4C 修饰在破骨细胞生成和炎性骨质流失中的调控机制。
方法
采用实时定量PCR(quantitative real-time PCR,qPCR)、western blotting、dot blot和免疫荧光染色法测定破骨细胞生成过程中NAT10的表达和ac4C修饰,采用抗酒石酸盐酸性磷酸酶染色、足小体带染色和骨吸收坑法测定NAT10抑制对破骨细胞分化的影响。然后,进行 acRIP-qPCR 和 NAT10RIP-qPCR、ac4C 位点预测、mRNA 衰变检测和荧光素酶报告基因分析,进一步研究其潜在机制。最后,应用炎症性骨质流失小鼠模型验证 NAT10 抑制在体内的治疗效果。
结果
NAT10 表达在破骨细胞分化过程中上调,在牙周炎小鼠的牙槽骨破骨细胞中高表达。抑制 NAT10 显着降低了体外破骨细胞的分化,表现为酒石酸盐耐药性酸性磷酸酶阳性多核细胞、破骨细胞特异性基因表达、F-肌动蛋白环形成和骨吸收能力的大幅减少。从机制上讲,NAT10 催化了 Fos (编码 AP-1 组分 c-Fos) mRNA 的 ac4C 修饰并保持其稳定性。此外,NAT10 促进 MAPK 信号通路,从而激活 AP-1 (c-Fos/c-Jun) 转录进行破骨细胞生成。在治疗上,NAT10 的特异性抑制剂 Remodelin 的给药显着阻碍了结扎诱导的牙槽骨流失和脂多糖诱导的炎性颅骨溶解。
结论
我们的研究表明,NAT10 介导的 ac4C 修饰是破骨细胞分化的重要表观遗传调控,并提出了一个有前途的炎症性骨质流失治疗靶点。






























 京公网安备 11010802027423号
京公网安备 11010802027423号