当前位置:
X-MOL 学术
›
Catal. Sci. Technol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Multistep screening of transition-metal-based homonuclear double-atom catalysts to unravel the electronic origin of their activity and selectivity challenges for nitrogen reduction
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2024-08-02 , DOI: 10.1039/d4cy00480a Anjumun Rasool 1 , Manzoor Ahmad Dar 1
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2024-08-02 , DOI: 10.1039/d4cy00480a Anjumun Rasool 1 , Manzoor Ahmad Dar 1
Affiliation
|
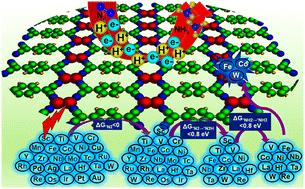
Lack of robust catalyst design strategies for tackling the selectivity and activity challenges poses serious limitations in the development of efficient catalysts for nitrogen reduction to ammonia. The synergistic interactions in double-atom catalysts (DACs) have aroused great interest in developing promising catalytic centers for the nitrogen reduction reaction (NRR). Using a multistep screening strategy based on systematic first-principles simulations, we find that Fe2, Co2, and W2 dimer species impregnated in a tetracyanoquinodimethane-based monolayer achieve suitable adsorption behaviour for the various NRR intermediates, leading to excellent activity and selectivity among the 27 DACs considered in this study for the NRR. Interestingly, our results reveal very low limiting potential values of −0.56, −0.58, and −0.53 V for Fe2, Co2, and W2, respectively, compared to the experimentally reported values of −0.73 and −0.98 V for the Ru-based single-atom catalyst and Ru(0001) stepped surface. Density of states analysis indicated that the adsorption pattern of the reaction intermediates was regulated by the d-states of the DACs near the Fermi level. Correlation trends between the limiting potential and the free energy change for adsorption of different intermediates show that the free energy change for N2 adsorption proves a suitable guidance to evaluate the NRR activity of the modelled catalysts. Further, rigorous electronic structure analysis highlighted properties such as integrated crystal orbital Hamilton populations and orbital projected density of states, and the d-band centre could be successfully used to rationalize the N2 binding and adsorption on these catalysts. Thus, this work provides a feasible design strategy for NRR electrocatalysis based on extensive electronic structure concepts.
中文翻译:

过渡金属基同核双原子催化剂的多步筛选,以揭示其活性的电子起源和氮还原的选择性挑战
缺乏应对选择性和活性挑战的稳健催化剂设计策略,严重限制了氮还原成氨的高效催化剂的开发。双原子催化剂(DAC)中的协同相互作用引起了人们对开发有前景的氮还原反应(NRR)催化中心的极大兴趣。采用基于系统第一原理模拟的多步筛选策略,我们发现浸渍在基于四氰基醌二甲烷的单层中的 Fe 2 、Co 2和 W 2二聚体物种对各种 NRR 中间体实现了合适的吸附行为,从而具有优异的活性和选择性在本研究中考虑的 NRR 的 27 个 DAC 中。有趣的是,我们的结果揭示了 Fe 2 、Co 2和 W 2的极限电势值非常低,分别为 -0.56、-0.58 和 -0.53 V,而 Ru 的实验报告值为 -0.73 和 -0.98 V。基于单原子催化剂和Ru(0001)阶梯表面。态密度分析表明反应中间体的吸附模式受到费米能级附近 DAC 的 d 态的调节。不同中间体吸附的极限电势和自由能变化之间的相关趋势表明,N 2吸附的自由能变化证明了评估模型催化剂的NRR活性的合适指导。 此外,严格的电子结构分析强调了积分晶体轨道汉密尔顿布居和轨道投影态密度等特性,并且d带中心可以成功地用于合理化这些催化剂上的N 2结合和吸附。因此,这项工作为基于广泛的电子结构概念的NRR电催化提供了可行的设计策略。
更新日期:2024-08-02
中文翻译:
过渡金属基同核双原子催化剂的多步筛选,以揭示其活性的电子起源和氮还原的选择性挑战
缺乏应对选择性和活性挑战的稳健催化剂设计策略,严重限制了氮还原成氨的高效催化剂的开发。双原子催化剂(DAC)中的协同相互作用引起了人们对开发有前景的氮还原反应(NRR)催化中心的极大兴趣。采用基于系统第一原理模拟的多步筛选策略,我们发现浸渍在基于四氰基醌二甲烷的单层中的 Fe 2 、Co 2和 W 2二聚体物种对各种 NRR 中间体实现了合适的吸附行为,从而具有优异的活性和选择性在本研究中考虑的 NRR 的 27 个 DAC 中。有趣的是,我们的结果揭示了 Fe 2 、Co 2和 W 2的极限电势值非常低,分别为 -0.56、-0.58 和 -0.53 V,而 Ru 的实验报告值为 -0.73 和 -0.98 V。基于单原子催化剂和Ru(0001)阶梯表面。态密度分析表明反应中间体的吸附模式受到费米能级附近 DAC 的 d 态的调节。不同中间体吸附的极限电势和自由能变化之间的相关趋势表明,N 2吸附的自由能变化证明了评估模型催化剂的NRR活性的合适指导。 此外,严格的电子结构分析强调了积分晶体轨道汉密尔顿布居和轨道投影态密度等特性,并且d带中心可以成功地用于合理化这些催化剂上的N 2结合和吸附。因此,这项工作为基于广泛的电子结构概念的NRR电催化提供了可行的设计策略。




































 京公网安备 11010802027423号
京公网安备 11010802027423号