Nature Genetics ( IF 31.7 ) Pub Date : 2024-07-29 , DOI: 10.1038/s41588-024-01842-3 Caroline F Wright 1 , Luke N Sharp 1 , Leigh Jackson 1 , Anna Murray 1 , James S Ware 2, 3, 4 , Daniel G MacArthur 5, 6 , Heidi L Rehm 4, 7 , Kashyap A Patel 1 , Michael N Weedon 1
|
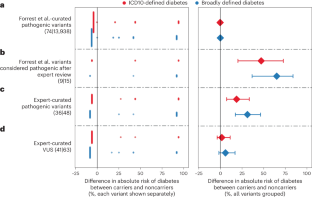
Penetrance is the probability that an individual with a pathogenic genetic variant develops a specific disease. Knowing the penetrance of variants for monogenic disorders is important for counseling of individuals. Until recently, estimates of penetrance have largely relied on affected individuals and their at-risk family members being clinically referred for genetic testing, a ‘phenotype-first’ approach. This approach substantially overestimates the penetrance of variants because of ascertainment bias. The recent availability of whole-genome sequencing data in individuals from very-large-scale population-based cohorts now allows ‘genotype-first’ estimates of penetrance for many conditions. Although this type of population-based study can underestimate penetrance owing to recruitment biases, it provides more accurate estimates of penetrance for secondary or incidental findings. Here, we provide guidance for the conduct of penetrance studies to ensure that robust genotypes and phenotypes are used to accurately estimate penetrance of variants and groups of similarly annotated variants from population-based studies.
中文翻译:
估计人群中单基因致病变异外显率的指南
外显率是具有致病性遗传变异的个体患特定疾病的概率。了解单基因疾病变异的外显率对于个人咨询很重要。直到最近,外显率的估计很大程度上依赖于受影响的个人及其高危家庭成员在临床上进行基因检测,这是一种“表型优先”的方法。由于确定偏差,这种方法大大高估了变异的外显率。最近,来自大规模人群的个体的全基因组测序数据的可用性现在允许对许多情况下的外显率进行“基因型优先”估计。尽管这种基于人群的研究可能由于招募偏差而低估外显率,但它为次要或偶然发现的外显率提供了更准确的估计。在这里,我们为进行外显率研究提供指导,以确保使用稳健的基因型和表型来准确估计基于人群的研究中的变异体和类似注释变异体组的外显率。










































 京公网安备 11010802027423号
京公网安备 11010802027423号