当前位置:
X-MOL 学术
›
J. Adv. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Cadmium targeting transcription factor EB to inhibit autophagy-lysosome function contributes to acute kidney injury
Journal of Advanced Research ( IF 11.4 ) Pub Date : 2024-07-19 , DOI: 10.1016/j.jare.2024.07.013 Peng-Fei Dong 1 , Tian-Bin Liu 2 , Kai Chen 2 , Dan Li 3 , Yue Li 1 , Cai-Yu Lian 1 , Zhen-Yong Wang 1 , Lin Wang 1
中文翻译:

靶向镉的转录因子 EB 抑制自噬溶酶体功能导致急性肾损伤
镉 (Cd) 的环境和职业暴露已被证明会导致急性肾损伤 (AKI)。既往研究表明,自噬抑制和溶酶体功能障碍是 Cd 诱导的 AKI 的重要机制。
转录因子 EB (TFEB) 是一种调节自噬溶酶体功能的关键转录调节因子,但其在 Cd 诱导的 AKI 中的作用尚不明确。因此,进行了体内和体外研究以阐明这个问题。
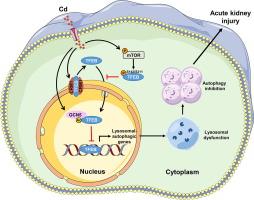
数据首先显示,在 Cd 诱导的 AKI 模型中,TFEB 表达降低和核转位明显,伴有自噬-溶酶体功能障碍。TFEB 的药理学和遗传激活通过减轻自噬抑制和溶酶体功能障碍改善 Cd 诱导的 AKI,而 Tfeb 敲除进一步加剧了这一现象,表明 TFEB 通过调节自噬在 Cd 诱导的 AKI 中起关键作用。从机制上讲,Cd 激活雷帕霉素复合物 1 (mTORC1) 的机制靶标以增强 TFEB 磷酸化,从而抑制 TFEB 核易位。Cd 还激活了染色体区域维持 1 (CRM1) 以促进 TFEB 核输出。同时,Cd 激活一般对照非抑制蛋白 5 (GCN5) 以增强核 TFEB 乙酰化,导致 TFEB 转录活性降低。此外,抑制 CRM1 或 GCN5 分别通过增强 TFEB 活性来减轻 Cd 诱导的 AKI。
综上所述,这些发现揭示了 TFEB 磷酸化、核输出和乙酰化通过调节自噬-溶酶体功能独立抑制 TFEB 活性导致 Cd 诱导的 AKI,表明 TFEB 激活可能是 Cd 诱导的 AKI 的一种有前途的治疗策略。
更新日期:2024-07-19
Journal of Advanced Research ( IF 11.4 ) Pub Date : 2024-07-19 , DOI: 10.1016/j.jare.2024.07.013 Peng-Fei Dong 1 , Tian-Bin Liu 2 , Kai Chen 2 , Dan Li 3 , Yue Li 1 , Cai-Yu Lian 1 , Zhen-Yong Wang 1 , Lin Wang 1
Affiliation
|
Introduction
Environmental and occupational exposure to cadmium (Cd) has been shown to cause acute kidney injury (AKI). Previous studies have demonstrated that autophagy inhibition and lysosomal dysfunction are important mechanisms of Cd-induced AKI.Objectives
Transcription factor EB (TFEB) is a critical transcription regulator that modulates autophagy-lysosome function, but its role in Cd-induced AKI is yet to be elucidated. Thus, in vivo and in vitro studies were conducted to clarify this issue.Methods and results
Data firstly showed that reduced TFEB expression and nuclear translocation were evident in Cd-induced AKI models, accompanied by autophagy-lysosome dysfunction. Pharmacological and genetic activation of TFEB improved Cd-induced AKI via alleviating autophagy inhibition and lysosomal dysfunction, whereas Tfeb knockdown further aggravated this phenomenon, suggesting the key role of TFEB in Cd-induced AKI by regulating autophagy. Mechanistically, Cd activated mechanistic target of rapamycin complex 1 (mTORC1) to enhance TFEB phosphorylation and thereby inhibiting TFEB nuclear translocation. Cd also activated chromosome region maintenance 1 (CRM1) to promote TFEB nuclear export. Meanwhile, Cd activated general control non-repressed protein 5 (GCN5) to enhance nuclear TFEB acetylation, resulting in the decreased TFEB transcriptional activity. Moreover, inhibition of CRM1 or GCN5 alleviated Cd-induced AKI by enhancing TFEB activity, respectively.Conclusion
In summary, these findings reveal that TFEB phosphorylation, nuclear export and acetylation independently suppress TFEB activity to cause Cd-induced AKI via regulating autophagy-lysosome function, suggesting that TFEB activation might be a promising treatment strategy for Cd-induced AKI.中文翻译:
靶向镉的转录因子 EB 抑制自噬溶酶体功能导致急性肾损伤
介绍
镉 (Cd) 的环境和职业暴露已被证明会导致急性肾损伤 (AKI)。既往研究表明,自噬抑制和溶酶体功能障碍是 Cd 诱导的 AKI 的重要机制。
目标
转录因子 EB (TFEB) 是一种调节自噬溶酶体功能的关键转录调节因子,但其在 Cd 诱导的 AKI 中的作用尚不明确。因此,进行了体内和体外研究以阐明这个问题。
方法和结果
数据首先显示,在 Cd 诱导的 AKI 模型中,TFEB 表达降低和核转位明显,伴有自噬-溶酶体功能障碍。TFEB 的药理学和遗传激活通过减轻自噬抑制和溶酶体功能障碍改善 Cd 诱导的 AKI,而 Tfeb 敲除进一步加剧了这一现象,表明 TFEB 通过调节自噬在 Cd 诱导的 AKI 中起关键作用。从机制上讲,Cd 激活雷帕霉素复合物 1 (mTORC1) 的机制靶标以增强 TFEB 磷酸化,从而抑制 TFEB 核易位。Cd 还激活了染色体区域维持 1 (CRM1) 以促进 TFEB 核输出。同时,Cd 激活一般对照非抑制蛋白 5 (GCN5) 以增强核 TFEB 乙酰化,导致 TFEB 转录活性降低。此外,抑制 CRM1 或 GCN5 分别通过增强 TFEB 活性来减轻 Cd 诱导的 AKI。
结论
综上所述,这些发现揭示了 TFEB 磷酸化、核输出和乙酰化通过调节自噬-溶酶体功能独立抑制 TFEB 活性导致 Cd 诱导的 AKI,表明 TFEB 激活可能是 Cd 诱导的 AKI 的一种有前途的治疗策略。






























 京公网安备 11010802027423号
京公网安备 11010802027423号