Nature ( IF 50.5 ) Pub Date : 2024-07-24 , DOI: 10.1038/s41586-024-07718-0 Juhyung Park , Jibo Wu , Krzysztof J. Szkop , Jinho Jeong , Predrag Jovanovic , Dylan Husmann , Natasha M. Flores , Joel W. Francis , Ying-Jiun C. Chen , Ana Morales Benitez , Emily Zahn , Shumei Song , Jaffer A. Ajani , Linghua Wang , Kamini Singh , Ola Larsson , Benjamin A. Garcia , Ivan Topisirovic , Or Gozani , Pawel K. Mazur
|
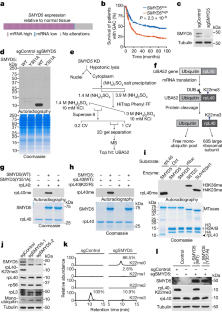
Dysregulated transcription due to disruption in histone lysine methylation dynamics is an established contributor to tumorigenesis1,2. However, whether analogous pathologic epigenetic mechanisms act directly on the ribosome to advance oncogenesis is unclear. Here we find that trimethylation of the core ribosomal protein L40 (rpL40) at lysine 22 (rpL40K22me3) by the lysine methyltransferase SMYD5 regulates mRNA translation output to promote malignant progression of gastric adenocarcinoma (GAC) with lethal peritoneal ascites. A biochemical–proteomics strategy identifies the monoubiquitin fusion protein partner rpL40 (ref. 3) as the principal physiological substrate of SMYD5 across diverse samples. Inhibiting the SMYD5–rpL40K22me3 axis in GAC cell lines reprogrammes protein synthesis to attenuate oncogenic gene expression signatures. SMYD5 and rpL40K22me3 are upregulated in samples from patients with GAC and negatively correlate with clinical outcomes. SMYD5 ablation in vivo in familial and sporadic mouse models of malignant GAC blocks metastatic disease, including peritoneal carcinomatosis. Suppressing SMYD5 methylation of rpL40 inhibits human cancer cell and patient-derived GAC xenograft growth and renders them hypersensitive to inhibitors of PI3K and mTOR. Finally, combining SMYD5 depletion with PI3K–mTOR inhibition and chimeric antigen receptor T cell administration cures an otherwise lethal in vivo mouse model of aggressive GAC-derived peritoneal carcinomatosis. Together, our work uncovers a ribosome-based epigenetic mechanism that facilitates the evolution of malignant GAC and proposes SMYD5 targeting as part of a potential combination therapy to treat this cancer.
中文翻译:
rpL40 的 SMYD5 甲基化将核糖体输出与胃癌联系起来
由于组蛋白赖氨酸甲基化动力学破坏而导致的转录失调是肿瘤发生的一个确定的因素 1,2 。然而,类似的病理表观遗传机制是否直接作用于核糖体以促进肿瘤发生尚不清楚。在这里,我们发现赖氨酸甲基转移酶 SMYD5 对核心核糖体蛋白 L40 (rpL40) 在赖氨酸 22 (rpL40K22me3) 上的三甲基化调节 mRNA 翻译输出,从而促进伴有致命腹膜腹水的胃腺癌 (GAC) 的恶性进展。生化-蛋白质组学策略将单泛素融合蛋白伴侣 rpL40(参考文献 3 )确定为不同样本中 SMYD5 的主要生理底物。抑制 GAC 细胞系中的 SMYD5–rpL40K22me3 轴可重新编程蛋白质合成,从而减弱致癌基因表达特征。 SMYD5 和 rpL40K22me3 在 GAC 患者样本中表达上调,并与临床结果呈负相关。在恶性 GAC 的家族性和散发性小鼠模型中体内 SMYD5 消除可阻止转移性疾病,包括腹膜癌病。抑制 rpL40 的 SMYD5 甲基化会抑制人类癌细胞和患者来源的 GAC 异种移植物的生长,并使它们对 PI3K 和 mTOR 抑制剂过敏。最后,将 SMYD5 耗竭与 PI3K-mTOR 抑制和嵌合抗原受体 T 细胞给药相结合,可以治愈一种致命的体内侵袭性 GAC 衍生腹膜癌病小鼠模型。我们的工作共同揭示了一种基于核糖体的表观遗传机制,该机制促进恶性 GAC 的进化,并提出 SMYD5 靶向作为治疗这种癌症的潜在联合疗法的一部分。











































 京公网安备 11010802027423号
京公网安备 11010802027423号