当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Modeling Intermolecular Coulombic Decay with Non-Hermitian Real-Time Time-Dependent Density Functional Theory
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2024-07-25 , DOI: 10.1021/acs.jpclett.4c01146 Yi-Siang Wang 1 , James X Zhong Manis 1 , Matthew C Rohan 1 , Thomas M Orlando 1 , Joshua S Kretchmer 1
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2024-07-25 , DOI: 10.1021/acs.jpclett.4c01146 Yi-Siang Wang 1 , James X Zhong Manis 1 , Matthew C Rohan 1 , Thomas M Orlando 1 , Joshua S Kretchmer 1
Affiliation
|
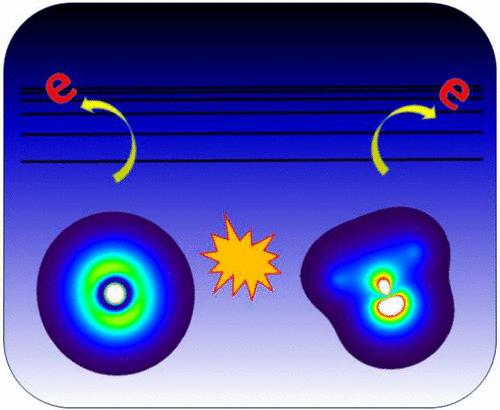
In this work, we investigate the capability of using real-time time-dependent density functional theory (RT-TDDFT) in conjunction with a complex absorbing potential (CAP) to simulate the intermolecular Coulombic decay (ICD) processes following the ionization of an inner-valence electron. We examine the ICD dynamics in a series of noncovalent bonded dimer systems, including hydrogen-bonded and purely van der Waals (VdW)-bonded systems. In comparison to previous work, we show that RT-TDDFT simulations with a CAP correctly capture the ICD phenomenon in systems exhibiting a stronger binding energy. The calculated time scales for ICD of the studied systems are in the range of 5–50 fs, in agreement with previous studies. However, there is a breakdown in the accuracy of the methodology for the pure VdW-bonded systems. Overall, the presented RT-TDDFT/CAP methodology provides a powerful tool for differentiating between competing electronic relaxation pathways following inner-valence or core ionization without necessitating any a priori assumptions.
中文翻译:

使用非厄米实时瞬态密度泛函理论模拟分子间库仑衰变
在这项工作中,我们研究了使用实时时间相关密度泛函理论 (RT-TDDFT) 与复杂吸收势 (CAP) 结合来模拟内部电离后的分子间库仑衰变 (ICD) 过程的能力。 -价电子。我们研究了一系列非共价键合二聚体系统中的 ICD 动力学,包括氢键和纯范德华 (VdW) 键合系统。与之前的工作相比,我们表明,使用 CAP 进行的 RT-TDDFT 模拟可以正确捕获表现出更强结合能的系统中的 ICD 现象。所研究系统的 ICD 计算时间尺度在 5-50 fs 范围内,与之前的研究一致。然而,纯 VdW 粘合系统的方法准确度有所下降。总的来说,所提出的 RT-TDDFT/CAP 方法提供了一个强大的工具,可以区分内价或核心电离后的竞争电子弛豫路径,而无需任何先验假设。
更新日期:2024-07-25
中文翻译:
使用非厄米实时瞬态密度泛函理论模拟分子间库仑衰变
在这项工作中,我们研究了使用实时时间相关密度泛函理论 (RT-TDDFT) 与复杂吸收势 (CAP) 结合来模拟内部电离后的分子间库仑衰变 (ICD) 过程的能力。 -价电子。我们研究了一系列非共价键合二聚体系统中的 ICD 动力学,包括氢键和纯范德华 (VdW) 键合系统。与之前的工作相比,我们表明,使用 CAP 进行的 RT-TDDFT 模拟可以正确捕获表现出更强结合能的系统中的 ICD 现象。所研究系统的 ICD 计算时间尺度在 5-50 fs 范围内,与之前的研究一致。然而,纯 VdW 粘合系统的方法准确度有所下降。总的来说,所提出的 RT-TDDFT/CAP 方法提供了一个强大的工具,可以区分内价或核心电离后的竞争电子弛豫路径,而无需任何先验假设。
















































 京公网安备 11010802027423号
京公网安备 11010802027423号