当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Correction to “Exploiting Molecular Symmetry to Quantitatively Map the Excited-State Landscape of Iron–Sulfur Clusters”
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2024-07-22 , DOI: 10.1021/jacs.4c08423 Brighton A. Skeel , Daniel L. M. Suess
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2024-07-22 , DOI: 10.1021/jacs.4c08423 Brighton A. Skeel , Daniel L. M. Suess
|
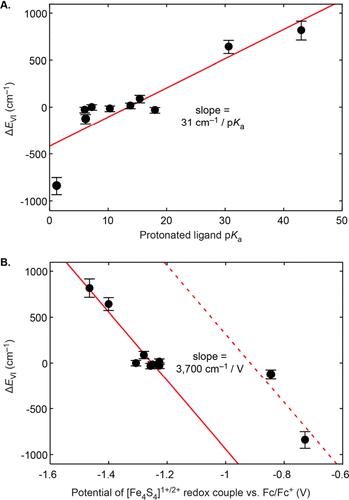
The fitting routine in the original article contains an incorrect formula for the solution magnetic moment. The correct expression (eq 1) is obtained by applying Boltzmann weights to the magnetization values of each spin state of the total spin ladder (1) and then converting this sum into an effective magnetic moment: The incorrect expression for magnetic moment utilized in the original article produced fitted magnetic moments that were systematically low in magnitude by approximately 15–25% for a given cluster over the temperature range sampled in our NMR experiments. Compensatorily, the original fitted superexchange couplings and valence isomer offset energies were generally too low in magnitude. As the corrected fits reported here show, the effect of this error was essentially a compression of the excited state manifolds for both valence isomer families and a slight shift and scaling in their offset energies relative to their correct values. Thus, although the fitted parameters reported in the original article are numerically incorrect, the qualitative magnetochemical trends observed for the series of clusters are unaltered. The corrected version of Figure 12 (page 10389 of the published article) shown herein illustrates this point, recapitulating the same correlations between ligand donor properties (pKa and [Fe4S4]1+/2+ redox potential) and ΔEVI that were identified in the original text, with the slopes of the lines of best fit being the only aspects of this figure that have meaningfully changed. Figure 12. Corrected version of Figure 12 in the original article. Linear best fits (solid red lines) have been recomputed for the corrected ΔEVI values. The dashed red trace in B) is equivalent in slope to the solid red trace but has been shifted by 0.34 V to empirically account for the shift in redox potential associated with the charge state change between the 1-X series and the [1-L]+ series. See the published article for the complete caption describing this figure. Corrected ΔEVI values are reported in Table 1 alongside their corresponding values as reported in the original text (page 10387 of the published article), for purposes of comparison. Other corrected fit parameters, corrected figures, and corrected MATLAB scripts for fitting VT NMR data are collected in the Supporting Information. Most differences between the original and corrected parameters are inconsequential and do not warrant further discussion. Here, we provided updated values for cases in which quantitative comparisons were explicitly made in the main text: In the original text, we estimated that substituting a thiolate ligand for an imidazole ligand yielded a ΔΔEVI value of ∼200 cm–1 (the original value of ΔΔEVI being ΔEVI(1-SBn) – ΔEVI([1-NMI]+) = 210 ± 60 cm–1). The corrected value for ΔΔEVI is unchanged (ΔΔEVI = 210 ± 60 cm–1), and we maintain that thiolate-for-imidazole substitutions change ΔEVI for [Fe4S4]1+ clusters by ∼200 cm–1. Whereas the originally reported total span in ΔEVI was ∼1000 cm–1, the corrected span is now ∼1700 cm–1, indicating that valence isomerism resulting from changes to the primary coordination sphere is at least as important for understanding the electronic structures of [Fe4S4]1+ clusters as we had previously concluded. Whereas in the original text we reported that global J values for the [Fe4S4]1+ state are on the order of 80–120 cm–1, we now find that these values are on the order of ∼200 cm–1 (corrected SI Table S1). Our originally reported value for Am(Fe2.5+) for the benzylic protons of 1-SBn was 0.98 MHz; the corrected value─1.45 MHz─is in better agreement with the literature value (2) of 1.32 MHz that was quoted in the original text. Thus, the primary conclusions of the original manuscript remain unaltered: For [Fe4S4]1+ clusters, a single ligand substitution can substantially perturb the relative energies of valence isomers, in some cases by ∼103 cm–1. More generally, applying symmetry considerations to synthetically modular clusters allows for the excited state landscape of Fe–S clusters to be quantitatively studied and systematically tuned. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c08423. Corrected fit parameters, other corrected figures, and corrected MATLAB scripts for fitting VT NMR data (PDF) Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html. This article references 2 other publications. This article has not yet been cited by other publications.
中文翻译:

对“利用分子对称性定量绘制铁硫簇的激发态景观”的修正
原文章中的拟合例程包含错误的磁矩解公式。正确的表达式(eq 1)是通过将玻尔兹曼权重应用于总自旋梯(1)的每个自旋状态的磁化值,然后将该总和转换为有效磁矩来获得的: 原始中使用的磁矩的错误表达式文章产生了拟合的磁矩,在我们 NMR 实验中采样的温度范围内,对于给定的簇,磁矩的大小系统地降低了约 15-25%。作为补偿,原来安装的超级交换耦合和化合价异构体偏移能的大小通常太低。正如此处报告的校正拟合所示,该误差的影响本质上是两个价异构体族的激发态流形的压缩以及它们的偏移能量相对于其正确值的轻微偏移和缩放。因此,尽管原始文章中报告的拟合参数在数值上不正确,但观察到的一系列簇的定性磁化学趋势并未改变。本文所示的图 12(已发表文章第 10389 页)的校正版本说明了这一点,概括了配体供体性质(p Ka和 [Fe 4 S 4 ] 1+/2+氧化还原电位)和 Δ E之间的相同相关性VI在原文中确定,最佳拟合线的斜率是该图唯一发生有意义变化的方面。图 12. 原始文章中图 12 的更正版本。 已针对校正的 Δ E VI值重新计算线性最佳拟合(红色实线)。 B) 中的红色虚线迹线的斜率与红色实线迹线相同,但已偏移 0.34 V,以根据经验解释与1-X系列和 [ 1-L之间的电荷态变化相关的氧化还原电位的变化] +系列。有关描述该图的完整标题,请参阅已发表的文章。校正后的 Δ E VI值与原文(已发表文章第 10387 页)中报告的相应值一起列于表 1 中,以供比较。用于拟合 VT NMR 数据的其他更正的拟合参数、更正的图形和更正的 MATLAB 脚本均收集在支持信息中。原始参数和校正参数之间的大多数差异都是无关紧要的,不需要进一步讨论。在这里,我们为正文中明确进行定量比较的情况提供了更新值:在原文中,我们估计用硫醇盐配体代替咪唑配体产生的 ΔΔ E VI值约为 200 cm –1 ( ΔΔ E VI的原始值为 Δ E VI ( 1-SBn ) – Δ E VI ([ 1-NMI ] + ) = 210 ± 60 cm –1 )。 ΔΔ E VI的校正值保持不变(ΔΔ E VI = 210 ± 60 cm –1 ),并且我们认为硫醇盐-咪唑取代使 [Fe 4 S 4 ] 1+簇的 Δ E VI变化约 200 cm –1 。最初报告的 Δ E VI总跨度为 ∼1000 cm –1 ,修正后的跨度现在为 ∼1700 cm –1 ,表明主配位层变化引起的价异构现象对于理解电子结构至少同样重要正如我们之前得出的结论,[Fe 4 S 4 ] 1+簇的数量。尽管在原文中我们报道了 [Fe 4 S 4 ] 1+态的全局J值约为 80–120 cm –1 ,但我们现在发现这些值约为 ∼200 cm –1 (更正了 SI 表 S1)。我们最初报告的1-SBn苄基质子的A m (Fe 2.5+ ) 值为 0.98 MHz;修正后的值──1.45 MHz──与原文引用的文献值(2)1.32 MHz较为吻合。因此,原始手稿的主要结论保持不变:对于[Fe 4 S 4 ] 1+簇,单个配体取代可以显着扰动价异构体的相对能量,在某些情况下约10 3 cm –1 。 更一般地说,将对称性考虑因素应用于合成模块化团簇可以对 Fe-S 团簇的激发态景观进行定量研究和系统调整。支持信息可在 https://pubs.acs.org/doi/10.1021/jacs.4c08423 免费获取。用于拟合 VT NMR 数据的更正的拟合参数、其他更正的图形和更正的 MATLAB 脚本 (PDF) 大多数电子支持信息文件无需订阅 ACS 网络版即可获得。此类文件可以按文章下载用于研究用途(如果有链接到相关文章的公共使用许可证,则该许可证可能允许其他用途)。可以通过 RightsLink 许可系统提出请求,从 ACS 获得许可用于其他用途:http://pubs.acs.org/page/copyright/permissions.html。本文引用了另外 2 篇出版物。这篇文章尚未被其他出版物引用。
更新日期:2024-07-22
中文翻译:
对“利用分子对称性定量绘制铁硫簇的激发态景观”的修正
原文章中的拟合例程包含错误的磁矩解公式。正确的表达式(eq 1)是通过将玻尔兹曼权重应用于总自旋梯(1)的每个自旋状态的磁化值,然后将该总和转换为有效磁矩来获得的: 原始中使用的磁矩的错误表达式文章产生了拟合的磁矩,在我们 NMR 实验中采样的温度范围内,对于给定的簇,磁矩的大小系统地降低了约 15-25%。作为补偿,原来安装的超级交换耦合和化合价异构体偏移能的大小通常太低。正如此处报告的校正拟合所示,该误差的影响本质上是两个价异构体族的激发态流形的压缩以及它们的偏移能量相对于其正确值的轻微偏移和缩放。因此,尽管原始文章中报告的拟合参数在数值上不正确,但观察到的一系列簇的定性磁化学趋势并未改变。本文所示的图 12(已发表文章第 10389 页)的校正版本说明了这一点,概括了配体供体性质(p Ka和 [Fe 4 S 4 ] 1+/2+氧化还原电位)和 Δ E之间的相同相关性VI在原文中确定,最佳拟合线的斜率是该图唯一发生有意义变化的方面。图 12. 原始文章中图 12 的更正版本。 已针对校正的 Δ E VI值重新计算线性最佳拟合(红色实线)。 B) 中的红色虚线迹线的斜率与红色实线迹线相同,但已偏移 0.34 V,以根据经验解释与1-X系列和 [ 1-L之间的电荷态变化相关的氧化还原电位的变化] +系列。有关描述该图的完整标题,请参阅已发表的文章。校正后的 Δ E VI值与原文(已发表文章第 10387 页)中报告的相应值一起列于表 1 中,以供比较。用于拟合 VT NMR 数据的其他更正的拟合参数、更正的图形和更正的 MATLAB 脚本均收集在支持信息中。原始参数和校正参数之间的大多数差异都是无关紧要的,不需要进一步讨论。在这里,我们为正文中明确进行定量比较的情况提供了更新值:在原文中,我们估计用硫醇盐配体代替咪唑配体产生的 ΔΔ E VI值约为 200 cm –1 ( ΔΔ E VI的原始值为 Δ E VI ( 1-SBn ) – Δ E VI ([ 1-NMI ] + ) = 210 ± 60 cm –1 )。 ΔΔ E VI的校正值保持不变(ΔΔ E VI = 210 ± 60 cm –1 ),并且我们认为硫醇盐-咪唑取代使 [Fe 4 S 4 ] 1+簇的 Δ E VI变化约 200 cm –1 。最初报告的 Δ E VI总跨度为 ∼1000 cm –1 ,修正后的跨度现在为 ∼1700 cm –1 ,表明主配位层变化引起的价异构现象对于理解电子结构至少同样重要正如我们之前得出的结论,[Fe 4 S 4 ] 1+簇的数量。尽管在原文中我们报道了 [Fe 4 S 4 ] 1+态的全局J值约为 80–120 cm –1 ,但我们现在发现这些值约为 ∼200 cm –1 (更正了 SI 表 S1)。我们最初报告的1-SBn苄基质子的A m (Fe 2.5+ ) 值为 0.98 MHz;修正后的值──1.45 MHz──与原文引用的文献值(2)1.32 MHz较为吻合。因此,原始手稿的主要结论保持不变:对于[Fe 4 S 4 ] 1+簇,单个配体取代可以显着扰动价异构体的相对能量,在某些情况下约10 3 cm –1 。 更一般地说,将对称性考虑因素应用于合成模块化团簇可以对 Fe-S 团簇的激发态景观进行定量研究和系统调整。支持信息可在 https://pubs.acs.org/doi/10.1021/jacs.4c08423 免费获取。用于拟合 VT NMR 数据的更正的拟合参数、其他更正的图形和更正的 MATLAB 脚本 (PDF) 大多数电子支持信息文件无需订阅 ACS 网络版即可获得。此类文件可以按文章下载用于研究用途(如果有链接到相关文章的公共使用许可证,则该许可证可能允许其他用途)。可以通过 RightsLink 许可系统提出请求,从 ACS 获得许可用于其他用途:http://pubs.acs.org/page/copyright/permissions.html。本文引用了另外 2 篇出版物。这篇文章尚未被其他出版物引用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号