Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Self-diffusion and shear viscosity of pure 1-alkanol unary system: molecular dynamics simulation and review of experimental data
RSC Advances ( IF 3.9 ) Pub Date : 2024-07-22 , DOI: 10.1039/d4ra03494e Adnan Jaradat 1 , Rakan Al-Salman 1 , Abdalla Obeidat 1
RSC Advances ( IF 3.9 ) Pub Date : 2024-07-22 , DOI: 10.1039/d4ra03494e Adnan Jaradat 1 , Rakan Al-Salman 1 , Abdalla Obeidat 1
Affiliation
|
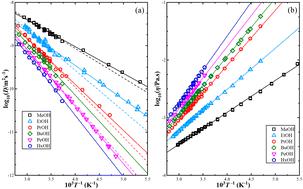
Self-diffusion coefficients and shear viscosity coefficients of pure 1-alkanol liquids from methanol to 1-hexanol were predicted using molecular dynamics (MD) simulations. These coefficients have been calculated using the Green–Kubo and Einstein methods at a range of temperatures of 200–330 K with increments of 10 K. Two force fields, TraPPE-UA and OPLS-AA were applied. The predicted results were compared to the experimental data, and the activation energies for self-diffusion and shear viscosity were calculated using the Arrhenius equation. The Stokes–Einstein equation was used to examine its capability in predicting the relationship between self-diffusion and shear viscosity, and the effective hydrodynamic radius was determined using both the experimental data and the results from MD simulations. The TraPPE-UA force field showed better results for the transport properties of methanol, while the OPLS-AA force field performed well for predicting shear viscosity but weakly for self-diffusion, particularly at low temperatures and for 1-alkanol with higher methylene numbers. Using the mean squared displacement method for self-diffusion was found to be more accurate than the Green–Kubo method, while the Green–Kubo method was slightly better for calculating shear viscosity. The Stokes–Einstein equation is valid for pure 1-alkanol liquids with temperature-dependent effective hydrodynamic radius.
中文翻译:

纯1-烷醇一元体系的自扩散和剪切粘度:分子动力学模拟和实验数据回顾
使用分子动力学 (MD) 模拟预测了从甲醇到 1-己醇的纯 1-烷醇液体的自扩散系数和剪切粘度系数。这些系数是使用 Green-Kubo 和 Einstein 方法在 200-330 K 的温度范围内以 10 K 的增量计算的。应用了两个力场:TraPPE-UA 和 OPLS-AA。将预测结果与实验数据进行比较,并使用阿伦尼乌斯方程计算自扩散活化能和剪切粘度。使用斯托克斯-爱因斯坦方程检验其预测自扩散和剪切粘度之间关系的能力,并使用实验数据和 MD 模拟结果确定有效流体动力学半径。 TraPPE-UA 力场在甲醇的传输特性方面表现出更好的结果,而 OPLS-AA 力场在预测剪切粘度方面表现良好,但在自扩散方面表现较弱,特别是在低温和具有较高亚甲基数的 1-烷醇时。发现使用均方位移法进行自扩散比 Green-Kubo 法更准确,而 Green-Kubo 法在计算剪切粘度方面稍好一些。斯托克斯-爱因斯坦方程对于具有与温度相关的有效流体动力学半径的纯 1-烷醇液体是有效的。
更新日期:2024-07-22
中文翻译:
纯1-烷醇一元体系的自扩散和剪切粘度:分子动力学模拟和实验数据回顾
使用分子动力学 (MD) 模拟预测了从甲醇到 1-己醇的纯 1-烷醇液体的自扩散系数和剪切粘度系数。这些系数是使用 Green-Kubo 和 Einstein 方法在 200-330 K 的温度范围内以 10 K 的增量计算的。应用了两个力场:TraPPE-UA 和 OPLS-AA。将预测结果与实验数据进行比较,并使用阿伦尼乌斯方程计算自扩散活化能和剪切粘度。使用斯托克斯-爱因斯坦方程检验其预测自扩散和剪切粘度之间关系的能力,并使用实验数据和 MD 模拟结果确定有效流体动力学半径。 TraPPE-UA 力场在甲醇的传输特性方面表现出更好的结果,而 OPLS-AA 力场在预测剪切粘度方面表现良好,但在自扩散方面表现较弱,特别是在低温和具有较高亚甲基数的 1-烷醇时。发现使用均方位移法进行自扩散比 Green-Kubo 法更准确,而 Green-Kubo 法在计算剪切粘度方面稍好一些。斯托克斯-爱因斯坦方程对于具有与温度相关的有效流体动力学半径的纯 1-烷醇液体是有效的。
















































 京公网安备 11010802027423号
京公网安备 11010802027423号