npj Parkinson's Disease ( IF 6.7 ) Pub Date : 2024-07-20 , DOI: 10.1038/s41531-024-00748-5
Madalynn L Erb 1, 2 , Kayla Sipple 1 , Nathan Levine 1 , Xi Chen 1 , Darren J Moore 1, 2
|
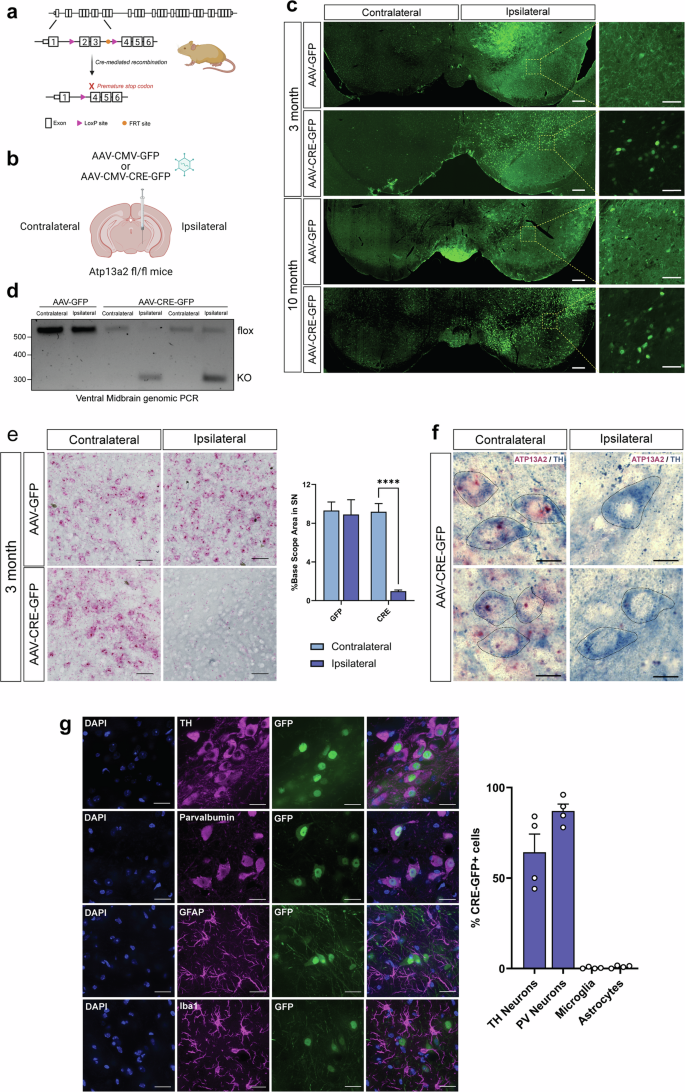
Although most cases of Parkinson’s disease (PD) are sporadic, mutations in over 20 genes are known to cause heritable forms of the disease. Recessive loss-of-function mutations in ATP13A2, a lysosomal transmembrane P5B-type ATPase and polyamine exporter, can cause early-onset familial PD. Familial ATP13A2 mutations are also linked to related neurodegenerative diseases, including Kufor-Rakeb syndrome, hereditary spastic paraplegias, neuronal ceroid lipofuscinosis, and amyotrophic lateral sclerosis. Despite the severe effects of ATP13A2 mutations in humans, ATP13A2 knockout (KO) mice fail to exhibit neurodegeneration even at advanced ages, making it challenging to study the neuropathological effects of ATP13A2 loss in vivo. Germline deletion of ATP13A2 in rodents may trigger the upregulation of compensatory pathways during embryonic development that mask the full neurotoxic effects of ATP13A2 loss in the brain. To explore this idea, we selectively deleted ATP13A2 in the adult mouse brain by the unilateral delivery of an AAV-Cre vector into the substantia nigra of young adult mice carrying conditional loxP-flanked ATP13A2 KO alleles. We observe a progressive loss of striatal dopaminergic nerve terminals at 3 and 10 months after AAV-Cre delivery. Cre-injected mice also exhibit robust dopaminergic neuronal degeneration in the substantia nigra at 10 months. Adult-onset ATP13A2 KO also recreates many of the phenotypes observed in aged germline ATP13A2 KO mice, including lysosomal abnormalities, p62-positive inclusions, and neuroinflammation. Our study demonstrates that the adult-onset homozygous deletion of ATP13A2 in the nigrostriatal pathway produces robust and progressive dopaminergic neurodegeneration that serves as a useful in vivo model of ATP13A2-related neurodegenerative diseases.
中文翻译:
小鼠成年发病的 ATP13A2 缺失可诱导进行性黑质纹状体通路多巴胺能变性和溶酶体异常
尽管大多数帕金森病 (PD) 病例是散发性的,但已知 20 多个基因的突变会导致该疾病的遗传形式。 ATP13A2 (一种溶酶体跨膜 P5 B型 ATP 酶和多胺输出蛋白)的隐性功能丧失突变可导致早发性家族性 PD。家族性ATP13A2突变也与相关的神经退行性疾病有关,包括 Kufor-Rakeb 综合征、遗传性痉挛性截瘫、神经元蜡质脂褐质沉着症和肌萎缩侧索硬化症。尽管ATP13A2突变对人类产生严重影响,但ATP13A2敲除 (KO) 小鼠即使在高龄也未能表现出神经退行性变,这使得在体内研究 ATP13A2 缺失的神经病理学影响具有挑战性。啮齿动物中ATP13A2的种系缺失可能会触发胚胎发育过程中补偿途径的上调,从而掩盖大脑中 ATP13A2 缺失的全部神经毒性作用。为了探索这个想法,我们通过将 AAV-Cre 载体单侧递送到携带条件性lox P 侧翼ATP13A2 KO 等位基因的年轻成年小鼠的黑质中,选择性地删除了成年小鼠大脑中的ATP13A2 。我们观察到 AAV-Cre 分娩后 3 个月和 10 个月时纹状体多巴胺能神经末梢逐渐丧失。注射 Cre 的小鼠在 10 个月时还表现出黑质中强烈的多巴胺能神经元变性。成年发病的ATP13A2 KO 还重现了在老年种系ATP13A2 KO 小鼠中观察到的许多表型,包括溶酶体异常、p62 阳性包涵体和神经炎症。 我们的研究表明,成年发病的黑质纹状体通路中ATP13A2纯合缺失会产生强烈且进行性的多巴胺能神经变性,可作为ATP13A2相关神经退行性疾病的有用体内模型。
































 京公网安备 11010802027423号
京公网安备 11010802027423号