Nature ( IF 50.5 ) Pub Date : 2024-07-17 , DOI: 10.1038/s41586-024-07707-3 Nicholas Morffy 1 , Lisa Van den Broeck 2 , Caelan Miller 1 , Ryan J Emenecker 3, 4 , John A Bryant 5 , Tyler M Lee 1 , Katelyn Sageman-Furnas 1 , Edward G Wilkinson 1 , Sunita Pathak 1 , Sanjana R Kotha 6 , Angelica Lam 6 , Saloni Mahatma 2 , Vikram Pande 2 , Aman Waoo 2 , R Clay Wright 5 , Alex S Holehouse 3, 4 , Max V Staller 6 , Rosangela Sozzani 2 , Lucia C Strader 1
|
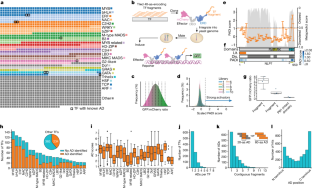
Gene expression in Arabidopsis is regulated by more than 1,900 transcription factors (TFs), which have been identified genome-wide by the presence of well-conserved DNA-binding domains. Activator TFs contain activation domains (ADs) that recruit coactivator complexes; however, for nearly all Arabidopsis TFs, we lack knowledge about the presence, location and transcriptional strength of their ADs1. To address this gap, here we use a yeast library approach to experimentally identify Arabidopsis ADs on a proteome-wide scale, and find that more than half of the Arabidopsis TFs contain an AD. We annotate 1,553 ADs, the vast majority of which are, to our knowledge, previously unknown. Using the dataset generated, we develop a neural network to accurately predict ADs and to identify sequence features that are necessary to recruit coactivator complexes. We uncover six distinct combinations of sequence features that result in activation activity, providing a framework to interrogate the subfunctionalization of ADs. Furthermore, we identify ADs in the ancient AUXIN RESPONSE FACTOR family of TFs, revealing that AD positioning is conserved in distinct clades. Our findings provide a deep resource for understanding transcriptional activation, a framework for examining function in intrinsically disordered regions and a predictive model of ADs.
中文翻译:
植物转录激活域的鉴定
拟南芥中的基因表达受到超过 1,900 个转录因子 (TF) 的调节,这些转录因子已在全基因组范围内通过存在高度保守的 DNA 结合域进行了鉴定。激活剂 TF 包含招募共激活剂复合物的激活域 (AD);然而,对于几乎所有拟南芥转录因子,我们缺乏对其 AD 的存在、位置和转录强度的了解1 。为了解决这一差距,我们使用酵母文库方法在蛋白质组范围内通过实验鉴定拟南芥AD,并发现超过一半的拟南芥TF 含有 AD。我们注释了 1,553 个公元元,据我们所知,其中绝大多数是以前未知的。使用生成的数据集,我们开发了一个神经网络来准确预测 AD 并识别招募共激活剂复合物所需的序列特征。我们发现了导致激活活性的序列特征的六种不同组合,提供了一个询问 AD 子功能化的框架。此外,我们在古老的生长素反应因子 TF 家族中鉴定了 AD,揭示了 AD 定位在不同的进化枝中是保守的。我们的研究结果为理解转录激活提供了深入的资源,为检查本质上无序区域的功能提供了框架,并提供了 AD 的预测模型。










































 京公网安备 11010802027423号
京公网安备 11010802027423号