当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Spin–Spin Coupling Constant Based on Reference Interaction Site Model Self-Consistent Field with Constrained Spatial Electron Density
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2024-07-15 , DOI: 10.1021/acs.jpclett.4c00948 Kosuke Imamura 1 , Daisuke Yokogawa 2 , Hirofumi Sato 1, 3
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2024-07-15 , DOI: 10.1021/acs.jpclett.4c00948 Kosuke Imamura 1 , Daisuke Yokogawa 2 , Hirofumi Sato 1, 3
Affiliation
|
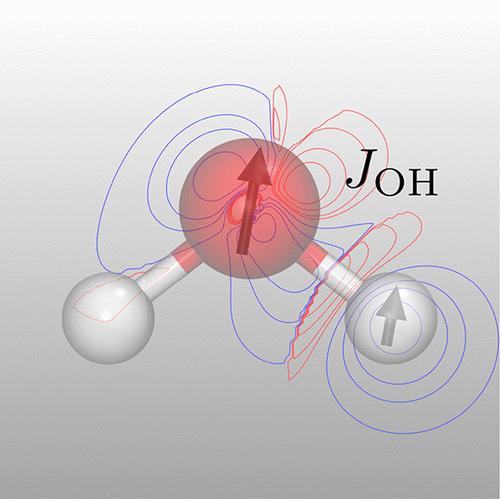
A method for computing spin–spin coupling constants (SSCCs) using the reference interaction site model self-consistent field with constrained spatial electron density (RISM-SCF-cSED) is proposed for the first time. Describing solvents using integral equation theory allows us to reflect solvent effects at atomic resolution in SSCCs while accounting for thermal fluctuations at a low computational cost. Applying the method to water, 1,1-difluoroethylene, and 1-methylaminomethylene-2-naphthalenone revealed that the solvent shift was evaluated to a greater extent than in the continuum solvent model. The origin of this phenomenon was analyzed in terms of the physical mechanisms underlying SSCCs.
中文翻译:

基于约束空间电子密度参考相互作用位点模型自洽场的自旋耦合常数
首次提出了一种利用空间电子密度受限的参考相互作用位点模型自洽场(RISM-SCF-cSED)计算自旋-自旋耦合常数(SSCC)的方法。使用积分方程理论描述溶剂使我们能够以原子分辨率反映 SSCC 中的溶剂效应,同时以较低的计算成本考虑热波动。将该方法应用于水、1,1-二氟乙烯和 1-甲基氨基亚甲基-2-萘酮表明,与连续溶剂模型相比,溶剂位移的评估程度更大。根据 SSCC 的物理机制分析了这种现象的起源。
更新日期:2024-07-15
中文翻译:
基于约束空间电子密度参考相互作用位点模型自洽场的自旋耦合常数
首次提出了一种利用空间电子密度受限的参考相互作用位点模型自洽场(RISM-SCF-cSED)计算自旋-自旋耦合常数(SSCC)的方法。使用积分方程理论描述溶剂使我们能够以原子分辨率反映 SSCC 中的溶剂效应,同时以较低的计算成本考虑热波动。将该方法应用于水、1,1-二氟乙烯和 1-甲基氨基亚甲基-2-萘酮表明,与连续溶剂模型相比,溶剂位移的评估程度更大。根据 SSCC 的物理机制分析了这种现象的起源。










































 京公网安备 11010802027423号
京公网安备 11010802027423号