当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Identification of novel LRRK2 inhibitors by structure-based virtual screening and alchemical free energy calculation
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-07-10 , DOI: 10.1039/d4cp01762e
Shuoyan Tan 1 , Xiaoqing Gong 2 , Huanxiang Liu 2 , Xiaojun Yao 2
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-07-10 , DOI: 10.1039/d4cp01762e
Shuoyan Tan 1 , Xiaoqing Gong 2 , Huanxiang Liu 2 , Xiaojun Yao 2
Affiliation
|
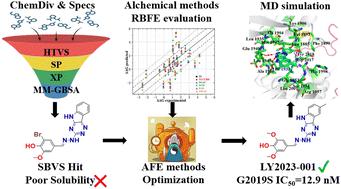
The Leucine-rich repeat kinase 2 (LRRK2) target has been identified as a promising drug target for Parkinson's disease (PD) treatment. This study focuses on optimizing the activity of LRRK2 inhibitors using alchemical relative binding free energy (RBFE) calculations. Initially, we assessed various free energy calculation methods across different LRRK2 kinase inhibitor scaffolds. The results indicate that alchemical free energy calculations are promising for prospective predictions on LRRK2 inhibitors, especially for the aminopyrimidine scaffold with an RMSE of 1.15 kcal mol−1 and Rp of 0.83. Following this, we optimized a potent LRRK2 kinase inhibitor identified from previous virtual screenings, featuring a novel scaffold. Guided by RBFE predictions using alchemical methods, this optimization led to the discovery of compound LY2023-001. This compound, with a [1,2,4]triazolo[5,6-b]indole scaffold, exhibited enhanced inhibitory activity against G2019S LRRK2 (IC50 = 12.9 nM). Molecular dynamics (MD) simulations revealed that LY2023-001 formed stable hydrogen bonds with Glu1948, and Ala1950 in the G2019S LRRK2 protein. Additionally, its phenyl substituents engage in strong electrostatic interactions with Lys1906 and van der Waals interactions with Leu1885, Phe1890, Val1893, Ile1933, Met1947, Leu1949, Leu2001, Ala2016, and Asp2017. Our findings underscore the potential of computational methods in the successful optimization of small molecules, offering important insights for the development of novel LRRK2 inhibitors.
中文翻译:

通过基于结构的虚拟筛选和炼金自由能计算鉴定新型 LRRK2 抑制剂
富含亮氨酸的重复激酶 2 (LRRK2) 靶点已被确定为帕金森病 (PD) 治疗的有前景的药物靶点。本研究的重点是使用炼金术相对结合自由能 (RBFE) 计算来优化 LRRK2 抑制剂的活性。最初,我们评估了不同 LRRK2 激酶抑制剂支架的各种自由能计算方法。结果表明,炼金自由能计算对于 LRRK2 抑制剂的前瞻性预测很有希望,特别是对于 RMSE 为 1.15 kcal mol -1和R p为 0.83 的氨基嘧啶支架。此后,我们优化了从之前的虚拟筛选中鉴定出的有效 LRRK2 激酶抑制剂,其具有新颖的支架。在使用炼金方法进行 RBFE 预测的指导下,这种优化导致了化合物LY2023-001的发现。该化合物具有[1,2,4]三唑并[5,6- b ]吲哚支架,对G2019S LRRK2表现出增强的抑制活性(IC 50 = 12.9 nM)。分子动力学(MD)模拟显示, LY2023-001与G2019S LRRK2蛋白中的Glu1948和Ala1950形成稳定的氢键。此外,其苯基取代基与 Lys1906 发生强静电相互作用,并与 Leu1885、Phe1890、Val1893、Ile1933、Met1947、Leu1949、Leu2001、Ala2016 和 Asp2017 发生范德华相互作用。我们的研究结果强调了计算方法在成功优化小分子方面的潜力,为新型 LRRK2 抑制剂的开发提供了重要的见解。
更新日期:2024-07-10
中文翻译:
通过基于结构的虚拟筛选和炼金自由能计算鉴定新型 LRRK2 抑制剂
富含亮氨酸的重复激酶 2 (LRRK2) 靶点已被确定为帕金森病 (PD) 治疗的有前景的药物靶点。本研究的重点是使用炼金术相对结合自由能 (RBFE) 计算来优化 LRRK2 抑制剂的活性。最初,我们评估了不同 LRRK2 激酶抑制剂支架的各种自由能计算方法。结果表明,炼金自由能计算对于 LRRK2 抑制剂的前瞻性预测很有希望,特别是对于 RMSE 为 1.15 kcal mol -1和R p为 0.83 的氨基嘧啶支架。此后,我们优化了从之前的虚拟筛选中鉴定出的有效 LRRK2 激酶抑制剂,其具有新颖的支架。在使用炼金方法进行 RBFE 预测的指导下,这种优化导致了化合物LY2023-001的发现。该化合物具有[1,2,4]三唑并[5,6- b ]吲哚支架,对G2019S LRRK2表现出增强的抑制活性(IC 50 = 12.9 nM)。分子动力学(MD)模拟显示, LY2023-001与G2019S LRRK2蛋白中的Glu1948和Ala1950形成稳定的氢键。此外,其苯基取代基与 Lys1906 发生强静电相互作用,并与 Leu1885、Phe1890、Val1893、Ile1933、Met1947、Leu1949、Leu2001、Ala2016 和 Asp2017 发生范德华相互作用。我们的研究结果强调了计算方法在成功优化小分子方面的潜力,为新型 LRRK2 抑制剂的开发提供了重要的见解。
































 京公网安备 11010802027423号
京公网安备 11010802027423号