Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2024-07-03 , DOI: 10.1002/adsc.202400517 Benoît CROUSSE 1 , murat sarac 2 , dimitra Kyrko 2 , pascal retailleau 3 , sandrine ongeri 4 , Jing HAO 4
|
The significant presence of fluorinated groups on organic compounds results in numerous applications in various fields, such as pharmaceuticals, agrochemicals and materials.1 For molecules exhibiting biological activities, fluorinated substituents improve their physicochemical properties (such as, for example, high lipophilicity, reduction in pKa, and modification of conformation), as well as their metabolic stability. Among the functions present in these molecules, α-(fluoroalkyl) amines combining a fluorinated functional group with a nitrogen heteroatom show interesting medicinal properties and biological activities (Figure 1).2

Some examples of bioactive trifluoromethyl amines.
Significant progress has been made in the formation of trifluoroethylamines2, 3 compared to the difluoroethylamines4 (Scheme 1).

Examples of other routes to α-fluorinated amines.
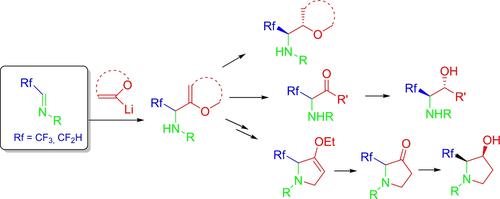
Among the various essential compounds used to synthesize fluoroalkyl amines, imines have emerged as powerful building blocks to construct novel fluorinated molecules.2a, 5 Unlike their nonfluorinated analogues, fluorinated imines are suitable electrophiles owing to the electron-withdrawing effect of the fluorinated group. Intensively involved in fluoroalkyl aldimine chemistry, our group is interested in their reactivity to access a wide range of functionalized trifluoromethyl amines, and in chiral forms. In this sense, the work undertaken has allowed the synthesis of α-trifluoromethylated allyl amines,6 homoallylic amines,7 and propargylic amines8 by the addition of organometallic reagents.
Vinyl ethers are found in many natural products and biologically active molecules. They represent one of the most reactive and synthetically valuable classes of hetero-substituted alkenes for synthesizing complex organic molecules. Employed as masked ketones and activated alkenes, vinyl ethers may be involved in many chemical transformations such as hydrolysis, reduction, cycloaddition reactions, Heck, and related cross-coupling reactions, including decarboxylative processes to produce poly-functionalized ketones, alcohols, heterocycles, and alkenes.9
To our knowledge, the addition of vinyl ether derivatives on trifluoromethyl and difluoromethyl aldimines and the transformation into functionalized fluorinated molecules were not reported in the literature. Thus far, we have focused on synthesizing α-trifluoromethyl and difluoromethyl amines using corresponding aldimines and vinyl ethers (Scheme 2).

Strategy to synthesize fluorinated amines.
N-p-Methoxyphenyl (PMP) and N-benzyl trifluoroacetaldimines 1 a,b were chosen as the model substrates for the reaction. First, nucleophilic organolithium reagent 2 a was prepared from ethyl vinyl ether with tert-butyl lithium (t-BuLi) in THF at −78 °C to 0 °C.10 Then, the lithiated ethyl vinyl ether 2 a was added to the imines 1 a,b at −78 °C. The reaction did not work with the imine 1 b, which decomposed in the media. Fortunately, adding the lithium reagent to the imine 1 a afforded the corresponding crude trifluoromethyl amine 3 a in excellent yield (Scheme 3).

Addition of vinyl ether on imine 1 a.
Owing to the high reactivity and the optimized conditions, we investigated the addition of other enol ethers on N-p-methoxyphenyl CF3 and HCF2 imines 1 a,c. Metalated ethyl vinyl ether 2 a, butyl vinyl ether 2 b, dihydrofuran 2 c, and dihydropyran 2 d react smoothly, affording solely products 3 in good to excellent yields (Table 1). Interestingly, the chiral CF3 sulfinylimine (S)-1 d reacted with lithiated ethyl and butyl vinyl ethers to afford amines 3 da and 3 db in 81% and 60% yields, respectively, and excellent diastereoselectivity (dr>98:2). From the dihydrofuran 2 c, the amine 3 dc was obtained in excellent yield but with a mixture of diastereomers (dr=85:15). From the chiral aldimine 1 e, the addition of the vinyl ether led to a low yield (14%) of expected compound 3 ea, with excellent diastereoselectivity (>98:2). However, a side product was isolated in 41% yield, but its structure was not elucidated. Considering the stereochemistry, the absolute configuration at the newly formed stereocenter of 3 da was assumed as S, according to the literature.11 The diastereoselectivity of alkyllithium additions can be rationalized by using a nonchelated model. The steric hindrance factors determine the selectivity. Alkyllithiums are preferably added to the imine from the less hindered face to afford the major diastereomer adducts (Ss,S)-3 da (Scheme 4).
|
|



Non-chelated transition state.
With several fluorinated enol ether amines in hand, we set out to study their reactivity under different conditions to access new functionalized fluorinated molecules. First, the double bond could be reduced under a hydrogen atmosphere with a catalytic amount of Pd/C (Scheme 5). Corresponding amino ethers 4 are obtained in excellent yields with a mixture of diastereomers,12 some of which can be easily separated by flash chromatography on silica gel. Fortunately, X-ray analyses of the major diastereomer of 4 ad were obtained and confirmed the anti configuration.

Reduction of vinyl ethers 3.
The hydrolysis of enol ethers was carried out in an acidic medium. Treating with a solution of 1 N HCl in THF under reflux for 1 h, the amino vinyl ethers 3 aa and 3 ab were wholly converted to the amino ketone 5 a. Due to its low stability,13 ketone 5 a was directly reduced with NaBH4 in MeOH provided amino alcohol 6 a as a mixture of diastereoisomers anti/syn (dr=95:5) in 57% yield (Scheme 6). This selectivity could be explained on one hand by the chelation of boron with the amine and the carbonyl, and on the other hand, by the attack of the hydride opposite to the CF3 group. Thanks to the achievement of the X-ray analysis of the major diastereomer, we confirm the anti stereochemistry (see S.I.).

Hydrolysis of CF3-amino vinyl ethers 3 and reduction of the ketone.
The same procedure was performed on the cyclic vinyl ethers 3 ac and 3 ad (Scheme 7). Ketones 5 were obtained in reasonable yields and directly reduced with NaBH4 to afford the corresponding amino alcohols 6 b and 6 c in 68% yield (dr=86:14) and 60% yield (dr=86:14), respectively. Major diastereomers obtained at the end of the reaction possess the anti stereochemistry according to the previous analysis of compound 6 a and the literature.12a Unfortunately, hydrolysis of the enantiomerically pure compound 3 da did not occur under these conditions, leading to compound mixtures, including racemization.

Hydrolysis of 3 and reduction of 5.
Another objective is to build new functionalized compounds from fluorinated amines with the enol ether function. After an N-allylation reaction, we attempted to prepare cyclic compounds by ring-closing metathesis (RCM). First, different conditions were tested using 3 aa to introduce the allyl group (Table 2). When the reaction proceeds with allyl bromide, NaHCO3, and KI in MeCN, the bis-allyl product 7 a is obtained in 50% yield after 10 days (Entry 1). The reaction with NaH in DMF didn't work, and the starting material remained (Entry 2). Using K2CO3 or NaHCO3 as a base in DMF, only a trace amount of product could be observed by 19F NMR after 2 days (Entries 3–4).
|
||||||
Entry |
Base |
Additive |
Solvent |
T(°C) |
Time (day) |
Yield (%) |
|---|---|---|---|---|---|---|
1 |
NaHCO3 |
KI |
MeCN |
100 |
10d |
50 |
2 |
NaH |
– |
DMF |
100 |
1d |
– |
3 |
K2CO3 |
– |
DMF |
100 |
2d |
Trace |
4 |
NaHCO3 |
KI |
DMF |
120 |
2d |
Trace |

Since the allylation reaction was too sluggish, a one-pot approach starting from imine was considered. The allyl group may be introduced more efficiently in a one-pot reaction due to the high reactivity of the lithium amide compound intermediate A. Thus, starting from CF3 and CF2H imines 1 a and 1 c, allyl bromide was added directly at −78 °C after adding lithiated vinyl ether 2 a. The allylation products 7 were obtained in 86% and 88% yields, respectively (Scheme 8). This one-pot reaction was fast, and starting materials were consumed completely.

Access to bis-unsaturated products 7 in one-pot approach.
In the same way, the vinylation and N-allylation reactions have been attempted on the chiral CF3 N-tert-butylsulfinyl ketimine 1 d. Unfortunately, the one pot on the sulfinylimines 1 d gave a lower yield. To circumvent failure, the N-allylation reaction was performed with allyl bromide and NaH in DMF14 with the sulfonamide 3 da’ previously oxidized with mCPBA. Thankfully, the final compound 7 d is isolated in good yield up to 71% (Scheme 9). Interestingly, the sulfonamide 3 da‘ crystallized, which made it possible to determine the configuration of the asymmetric center; the configuration is S.

Access to chiral bis-unsaturated products.
Concerning the RCM reaction of vinyl ether and alkene, a few examples are reported in the literature using the Grubbs 2nd catalyst.9d, 15 We attempted the RCM reaction with 5 mol% Grubbs 2nd on the bis-unsaturated compounds 7. The reaction proceeded at reflux of toluene for 2 h to give the functionalized cyclic fluorinated amines 9 in good yields (Scheme 10).

RCM reaction on 7.
The compounds 9 could be an important platform for constructing interestingly functionalized cyclic molecules. Different conditions have been attempted to functionalize the double bond. First, the hydrogenation was carried out under hydrogen at room temperature with Pd/C catalyst. The corresponding saturated compounds 10–11 were obtained in reasonable yields and excellent diastereoselectivity. Next, the hydrolysis under acid conditions (HCl) afforded ketones, which are directly reduced by NaBH4 in methanol. Here also, compounds 12–13 were isolated in good yields and excellent stereoselectivities. The majority configuration of the compounds 10–13 is Cis due to the hindrance of the CF3 group on the cycles (Scheme 11). X-rays of the majority of compound 12 confirm this stereochemistry.

Some transformations of the compounds 9.
We have attempted deprotection of the chiral group, but the main conditions are strong acidic conditions incompatible with vinyl ethers. As a result, the deprotection of the chiral group as well as the hydrolysis of the vinyl ether and the presence of several products, are observed. So, the deprotection works, but it would be more reasonable to perform deprotection on chiral compounds once the vinyl ether functions are no longer present. In addition, the complete deprotection of the N-PMP protecting group could be carried out, for example, on compound 3 ca in the presence of ceric ammonium nitrate (CAN) to lead to amine 14 without altering the vinyl ether function (Scheme 12).

Deprotection of the PMP group.
In conclusion, we have disclosed the addition reaction of lithium enol ether reagents to fluorinated aldimines and chiral fluorinated N-tert-butylsulfinylketimines. Various CF3 and CF2H amines were obtained, including β-amino ketones, β-amino alcohols, and cyclic amines, which are difficult to obtain by other methods. This selective addition reaction provided a straightforward process for synthesizing enantiomerically enriched α-fluorinated amines and derivatives. Good diastereoselectivities were achieved, and the corresponding products were obtained in good to excellent yields. These preliminary results encourage exemplifying the reactivity of these fluorinated amino-enol ethers in different fields.
Experimental Section
Typical Procedure for the Addition of Lithium Enol Ether 2 a to Fluoroalkyl Imine 1 a
At a solution of vinyl ether (3 eq.) dissolved in THF (25 mL) under Ar atmosphere. Then t-BuLi (2 eq.) was added dropwise at −78 °C, and the reaction mixture let stirring at 0 °C for 1 h. After the reaction was cooled again at −78 °C, and the imine (1 eq.) was added dropwise. Back to 0°C and after 2 h, the reaction mixture was treated by saturated aqueous solution of NH4Cl, extracted with Et2O two times, dried over MgSO4, concentrated under vacuum. The product was obtained as brown oil in 77% yield.
CCDC-2352969 (3 da′), −2352970 (6 a), −2352971 (4 ad), and −2364944 (12) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
Acknowledgments
The Ministère de l'Enseignement Supérieur et de la Recherche (MESR) is thanked for financial support for Dimitra Kyrko. The authors gratefully acknowledge Central Glass Co. for the gift of trifluoro and difluoroacetaldehyde hemiacetal. The French Fluorine Network (GIS-FLUOR) is also acknowledged for its support.
中文翻译:
来自亚胺和烯醇醚的官能化 α-氟化胺
有机化合物上大量存在的氟化基团导致其在各个领域的广泛应用,例如药物、农用化学品和材料。 1对于表现出生物活性的分子,氟化取代基可改善其理化性质(例如,高亲脂性、pKa 降低和构象修饰)及其代谢稳定性。在这些分子中存在的功能中,结合氟化官能团与氮杂原子的 α-(氟烷基)胺显示出有趣的药用特性和生物活性(图 1)。 2
在图查看器PowerPoint中打开
生物活性三氟甲基胺的一些例子。
与二氟乙胺4相比,三氟乙胺2、3的形成取得了重大进展(方案 1)。
在图查看器PowerPoint中打开
α-氟化胺的其他途径的例子。
在用于合成氟烷基胺的各种重要化合物中,亚胺已成为构建新型氟化分子的强大构建单元。 2a, 5与非氟化类似物不同,氟化亚胺由于氟化基团的吸电子效应而成为合适的亲电子试剂。我们的团队深入参与氟烷基醛亚胺化学,对其获得各种手性形式的官能化三氟甲基胺的反应性感兴趣。从这个意义上说,所开展的工作允许通过添加有机金属试剂来合成α-三氟甲基化烯丙胺、 6高烯丙胺7和炔丙胺8 。
乙烯基醚存在于许多天然产物和生物活性分子中。它们代表了用于合成复杂有机分子的最具反应性和合成价值的杂取代烯烃类别之一。乙烯基醚用作掩蔽酮和活化烯烃,可参与许多化学转化,例如水解、还原、环加成反应、Heck 和相关交叉偶联反应,包括脱羧过程以产生多官能化酮、醇、杂环和烯烃。 9
据我们所知,文献中尚未报道在三氟甲基和二氟甲基醛亚胺上添加乙烯基醚衍生物以及转化为官能化氟化分子。到目前为止,我们的重点是使用相应的醛亚胺和乙烯基醚合成α-三氟甲基胺和二氟甲基胺(方案2)。
在图查看器PowerPoint中打开
氟化胺的合成策略。
选择N-对甲氧基苯基(PMP)和N-苄基三氟乙醛亚胺1a 、 b作为反应的模型底物。首先,在-78℃至0℃下,在THF中,由乙基乙烯基醚与叔丁基锂( t- BuLi)制备亲核有机锂试剂2a 。 10然后,在-78℃下将锂化乙基乙烯基醚2a添加到亚胺1a 、 b中。该反应不适用于亚胺1 b ,它在介质中分解。幸运的是,将锂试剂添加到亚胺1a 中,以优异的收率得到了相应的粗三氟甲基胺3a (方案 3)。
在图查看器PowerPoint中打开
乙烯基醚在亚胺上加成1a 。
由于其高反应活性和优化的条件,我们研究了其他烯醇醚在N-对甲氧基苯基CF 3和HCF 2亚胺1 a , c上的加成反应。金属化乙基乙烯基醚2a 、丁基乙烯基醚2b 、二氢呋喃2c和二氢吡喃2d反应顺利,仅以良好至优异的收率得到产物3 (表1)。有趣的是,手性CF 3亚磺酰亚胺( S ) -1d与锂化乙基和丁基乙烯基醚反应得到胺3da和3db,产率分别为81%和60%,并且具有优异的非对映选择性(dr>98:2)。从二氢呋喃2c ,以优异的产率获得胺3dc ,但具有非对映异构体的混合物(dr=85:15)。从手性醛亚胺1 e中,添加乙烯基醚得到了低产率 (14%) 的预期化合物3 ea ,且具有优异的非对映选择性 (>98:2)。然而,以 41% 的产率分离出副产物,但其结构尚未阐明。根据文献,考虑到立体化学,新形成的3 da立体中心的绝对构型被假定为S 。 11烷基锂加成的非对映选择性可以通过使用非螯合模型合理化。位阻因素决定了选择性。优选将烷基锂从受阻较小的面添加到亚胺中,以提供主要的非对映异构体加合物( Ss,S ) -3da (方案4)。
|
|
在图查看器PowerPoint中打开
非螯合过渡态。
有了几种氟化烯醇醚胺,我们开始研究它们在不同条件下的反应性,以获得新的官能化氟化分子。首先,双键可以在氢气气氛下用催化量的 Pd/C 还原(方案 5)。相应的氨基醚4以优异的产率与非对映异构体的混合物获得,其中一些可以通过硅胶快速色谱法轻松分离。幸运的是,获得了4 ad主要非对映异构体的 X 射线分析并证实了反构型。
在图查看器PowerPoint中打开
乙烯基醚的还原3 .
烯醇醚的水解在酸性介质中进行。用1N HCl的THF溶液回流处理1小时,氨基乙烯基醚3aa和3ab完全转化为氨基酮5a 。由于稳定性较低, 13酮5a直接用 NaBH 4在 MeOH 中还原,得到氨基醇6a,为非对映异构体anti/syn (dr=95:5) 的混合物,产率 57%(方案 6)。这种选择性一方面可以通过硼与胺和羰基的螯合来解释,另一方面可以通过与CF 3基团相反的氢化物的攻击来解释。由于主要非对映异构体的 X 射线分析的实现,我们确认了反立体化学(参见 SI)。
在图查看器PowerPoint中打开
CF 3 -氨基乙烯基醚3的水解和酮的还原。
对环状乙烯基醚3 ac和3 ad进行相同的程序(方案 7)。以合理的收率获得酮5 ,并直接用NaBH 4还原,分别以68%收率(dr=86:14)和60%收率(dr=86:14)得到相应的氨基醇6b和6c 。根据先前对化合物6a的分析和文献,反应结束时获得的主要非对映异构体具有反立体化学。 12a不幸的是,对映体纯的化合物3 da在这些条件下没有发生水解,导致化合物混合物,包括外消旋化。
在图查看器PowerPoint中打开
3的水解和5的还原。
另一个目标是从具有烯醇醚功能的氟化胺构建新的功能化化合物。经过N-烯丙基化反应后,我们尝试通过闭环复分解(RCM)制备环状化合物。首先,使用3 个氨基酸测试不同条件引入烯丙基(表 2)。当反应与烯丙基溴、NaHCO 3和KI在MeCN中进行时,10天后以50%的产率获得双烯丙基产物7a (条目1)。与 DMF 中的 NaH 的反应不起作用,并且保留了起始材料(条目 2)。使用 K 2 CO 3或 NaHCO 3作为 DMF 中的碱,2 天后19 F NMR 只能观察到痕量产物(条目 3-4)。
表2.与烯丙基溴的N-烯丙基化反应。
|
||||||
入口 |
根据 |
添加剂 |
溶剂 |
温度(℃) |
时间(天) |
屈服 (%) |
|---|---|---|---|---|---|---|
1 |
碳酸氢钠3 |
KI |
甲基氰 |
100 |
10天 |
50 |
2 |
氢化钠 |
– |
DMF |
100 |
1天 |
– |
3 |
K2CO3 |
– |
DMF |
100 |
2d |
痕迹 |
4 |
碳酸氢钠3 |
KI |
DMF |
120 |
2d |
痕迹 |
由于烯丙基化反应太慢,因此考虑从亚胺开始的一锅法。由于氨基锂化合物中间体A的高反应性,可以在一锅反应中更有效地引入烯丙基。因此,从CF 3和CF 2 H亚胺1a和1c开始,在添加锂化乙烯基醚2a之后,在-78℃下直接添加烯丙基溴。烯丙基化产物7的产率分别为 86% 和 88%(方案 8)。这种一锅反应速度快,起始原料被完全消耗。
在图查看器PowerPoint中打开
以一锅法获得双不饱和产物7 。
以同样的方式,对手性CF 3 N-叔丁基亚磺酰基酮亚胺1d进行了乙烯基化和N-烯丙基化反应。不幸的是,一锅亚磺酰亚胺1天的产率较低。为了避免失败,用烯丙基溴和NaH在DMF 14中与预先用mCPBA氧化的磺酰胺3da'进行N-烯丙基化反应。值得庆幸的是,最终化合物7d的分离收率高达 71%(方案 9)。有趣的是,磺酰胺3 da'结晶,这使得确定不对称中心的构型成为可能;配置为S 。
在图查看器PowerPoint中打开
获得手性双不饱和产品。
关于乙烯基醚和烯烃的RCM反应,文献中报道了一些使用Grubbs 2nd催化剂的例子。 9d, 15我们尝试用 5 mol% Grubbs 2 nd对双不饱和化合物7进行 RCM 反应。反应在甲苯回流下进行2小时,以良好的收率得到官能化的环状氟化胺9 (方案10)。
在图查看器PowerPoint中打开
RCM 反应于7 。
化合物9可能是构建有趣的功能化环状分子的重要平台。已尝试不同的条件来使双键官能化。首先,在氢气环境下,使用Pd/C催化剂,在室温下进行氢化反应。以合理的产率和优异的非对映选择性获得了相应的饱和化合物10 – 11 。接下来,在酸性条件(HCl)下水解得到酮,其在甲醇中被NaBH 4直接还原。在此,化合物12 – 13也以良好的收率和优异的立体选择性被分离出来。由于 CF 3基团对循环的阻碍,化合物10 – 13的大多数构型是顺式(方案 11)。大部分化合物12的 X 射线证实了这种立体化学。
在图查看器PowerPoint中打开
化合物9的一些转变。
我们尝试了手性基团的脱保护,但主要条件是与乙烯基醚不相容的强酸性条件。结果,观察到手性基团的脱保护以及乙烯基醚的水解和多种产物的存在。因此,脱保护是有效的,但一旦乙烯基醚官能团不再存在,对手性化合物进行脱保护会更合理。此外,可以在硝酸高铈铵 (CAN) 存在下对化合物3 ca进行N -PMP 保护基的完全脱保护,得到胺14 ,而不改变乙烯基醚官能团(方案 12) 。
在图查看器PowerPoint中打开
PMP 基团的脱保护。
总之,我们公开了锂烯醇醚试剂与氟化醛亚胺和手性氟化N-叔丁基亚磺酰基酮亚胺的加成反应。获得了多种CF 3和CF 2 H胺,包括β-氨基酮、β-氨基醇和环胺,这是其他方法难以获得的。这种选择性加成反应提供了合成对映体富集的 α-氟化胺和衍生物的简单方法。实现了良好的非对映选择性,并以良好至优异的收率获得了相应的产物。这些初步结果鼓励举例说明这些氟化氨基烯醇醚在不同领域的反应活性。
实验部分
将锂烯醇醚 2a 加成至氟代烷基亚胺 1a 的典型程序
在Ar气氛下,将乙烯基醚(3当量)溶解在THF(25mL)中。然后在-78℃下滴加t -BuLi(2当量),并将反应混合物在0℃下搅拌1小时。将反应物再次冷却至-78℃后,滴加亚胺(1当量)。回到0℃并在2小时后,将反应混合物用饱和NH 4 Cl水溶液处理,用Et 2 O萃取两次,经MgSO 4干燥,真空浓缩。获得棕色油状产物,产率77%。
CCDC-2352969 ( 3 da′ )、-2352970 ( 6 a )、-2352971 ( 4 ad ) 和 -2364944 ( 12 ) 包含本文的补充晶体学数据。这些数据可以通过 www.ccdc.cam.ac.uk/structs 从剑桥晶体学数据中心免费获得。
致谢
感谢 Ministère de l'Enseignement Supérieur et de la Recherche (MESR) 对 Dimitra Kyrko 的财政支持。作者衷心感谢中央玻璃公司赠送的三氟乙醛和二氟乙醛半缩醛。法国氟网络 (GIS-FLUOR) 的支持也得到认可。










































 京公网安备 11010802027423号
京公网安备 11010802027423号