npj Computational Materials ( IF 9.4 ) Pub Date : 2024-07-02 , DOI: 10.1038/s41524-024-01321-7 Josiah Roberts , Biswas Rijal , Simon Divilov , Jon-Paul Maria , William G. Fahrenholtz , Douglas E. Wolfe , Donald W. Brenner , Stefano Curtarolo , Eva Zurek
|
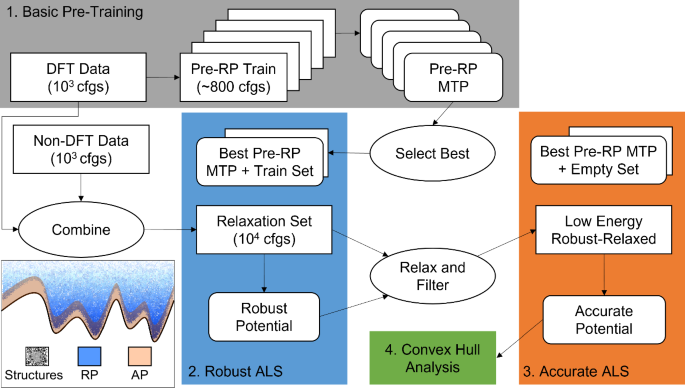
Large-density functional theory (DFT) databases are a treasure trove of energies, forces, and stresses that can be used to train machine-learned interatomic potentials for atomistic modeling. Herein, we employ structural relaxations from the AFLOW database to train moment tensor potentials (MTPs) for four carbide systems: CHfTa, CHfZr, CMoW, and CTaTi. The resulting MTPs are used to relax ~6300 random symmetric structures, and are subsequently improved via active learning to generate robust potentials (RP) that can relax a wide variety of structures, and accurate potentials (AP) designed for the relaxation of low-energy systems. This protocol is shown to yield convex hulls that are indistinguishable from those predicted by AFLOW for the CHfTa, CHfZr, and CTaTi systems, and in the case of the CMoW system to predict thermodynamically stable structures that are not found within AFLOW, highlighting the potential of the employed protocol within crystal structure prediction. Relaxation of over three hundred (Mo1−xWx)C stoichiometry crystals first with the RP then with the AP yields formation enthalpies that are in excellent agreement with those obtained via DFT.
中文翻译:
在 AFLOW 数据库上训练的机器学习三元碳化物原子间势
大密度泛函理论 (DFT) 数据库是能量、力和应力的宝库,可用于训练机器学习的原子间势以进行原子建模。在这里,我们利用 AFLOW 数据库中的结构松弛来训练四种碳化物系统的矩张量势 (MTP):CHfTa、CHfZr、CMoW 和 CTaTi。由此产生的 MTP 用于弛豫约 6300 个随机对称结构,并随后通过主动学习进行改进,以生成可以弛豫各种结构的鲁棒电位 (RP),以及专为弛豫低能量结构而设计的精确电位 (AP)系统。该协议显示出对于 CHfTa、CHfZr 和 CTaTi 系统产生的凸包与 AFLOW 预测的凸包没有区别,并且在 CMoW 系统的情况下,可以预测 AFLOW 中未发现的热力学稳定结构,这凸显了晶体结构预测中使用的协议。首先用 RP 然后用 AP 对超过 300 个 (Mo 1−x W x )C 化学计量晶体进行弛豫,产生的生成焓与通过 DFT 获得的结果非常一致。











































 京公网安备 11010802027423号
京公网安备 11010802027423号