Molecular Diversity ( IF 3.9 ) Pub Date : 2024-07-02 , DOI: 10.1007/s11030-024-10916-7 Tianli Qin 1, 2 , Yijian Wang 1 , Miaomiao Kong 1 , Hongliang Zhong 1, 3 , Tao Wu 1 , Zixuan Xi 1 , Zhenyong Qian 1, 3 , Ke Li 1, 3 , Yuepiao Cai 4 , Jianzhang Wu 2, 3 , Wulan Li 1
|
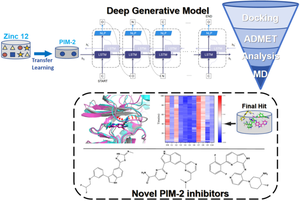
Proviral Integrations of Moloney-2 (PIM-2) kinase is a promising target for various cancers and other diseases, and its inhibitors hold potential for treating related diseases. However, there is currently no clinically available PIM-2 inhibitor. In this study, we constructed a generative model for de novo PIM-2 inhibitor design based on artificial intelligence, performed molecular docking and molecular dynamics (MD) simulations to develop an efficient PIM-2 inhibitor generative model and discover potential PIM-2 inhibitors. First, we designed a generative model based on a Bi-directional Long Short-Term Memory (BiLSTM) framework combined with a transfer learning strategy and generated a new PIM-2 small molecule library using existing active drug databases. The generated compound library was then virtually screened by molecular docking and scaffold similarity comparison, identifying 10 initial hit compounds with better performance. Next, using the inhibitor in the crystal structure as a positive control, we performed two rounds of MD simulations, with lengths of 100 ns and 500 ns, respectively, to study the dynamic stability of the protein–ligand systems of the 10 compounds with PIM-2. Analyzed the interactions with key hinge region residues, binding free energies, and changes in the ATP pocket size. The generative model demonstrates good molecular generation capability and can generate efficient novel molecules with similar physicochemical properties as active PIM-2 drugs. Among the 10 initially selected hit compounds, 5 compounds C3 (− 29.69 kcal/mol), C4 (− 33.31 kcal/mol), C5 (− 28.59 kcal/mol), C8 (− 34.68 kcal/mol), and C9 (− 25.88 kcal/mol) have higher binding energies with PIM-2 than the positive drug 3YR (− 26.18 kcal/mol). The MD simulation results are consistent with the docking analysis, these compounds have lower and more stable RMSD values for the complex systems with the reported positive drug 3YR and PIM-2 complex system. They can form long-term stable interactions with active site and the hinge region of PIM-2, which suggests these compounds are likely to have potent inhibitory effects on PIM-2. This study provides an efficient generative model for PIM-2 inhibitor research and discovers 5 potential novel PIM-2 inhibitors.
Graphical abstract
中文翻译:
通过基于配体的生成模型、分子对接和分子动力学模拟鉴定潜在的 PIM-2 抑制剂
Moloney-2 (PIM-2) 激酶的前病毒整合是各种癌症和其他疾病的一个有前途的靶点,其抑制剂具有治疗相关疾病的潜力。然而,目前尚无临床可用的 PIM-2 抑制剂。在这项研究中,我们基于人工智能构建了从头 PIM-2 抑制剂设计的生成模型,进行了分子对接和分子动力学 (MD) 模拟,以开发高效的 PIM-2 抑制剂生成模型并发现潜在的 PIM-2 抑制剂。首先,我们设计了一个基于双向长短期记忆 (BiLSTM) 框架的生成模型,并结合了迁移学习策略,并使用现有的活性药物数据库生成了一个新的 PIM-2 小分子库。然后通过分子对接和支架相似性比较对生成的化合物库进行虚拟筛选,鉴定出 10 个性能更好的初始命中化合物。接下来,使用晶体结构中的抑制剂作为阳性对照,我们进行了两轮 MD 模拟,长度分别为 100 ns 和 500 ns,以研究 PIM-2 的 10 种化合物的蛋白质-配体系统的动态稳定性。分析了与关键铰链区残基、结合自由能和 ATP 口袋大小变化的相互作用。生成模型表现出良好的分子生成能力,可以生成与活性 PIM-2 药物具有相似理化性质的高效新分子。在最初选择的 10 种命中化合物中,5 种化合物 C3 (- 29.69 kcal/mol)、C4 (- 33.31 kcal/mol)、C5 (-28.59 kcal/mol)、C8 (-34.68 kcal/mol) 和 C9 (-25.88 kcal/mol) 与 PIM-2 的结合能高于阳性药物 3YR (- 26.18 kcal/mol)。 MD 模拟结果与对接分析一致,对于已报道的阳性药物 3YR 和 PIM-2 复合物系统的复杂系统,这些化合物具有更低且更稳定的 RMSD 值。它们可以与 PIM-2 的活性位点和铰链区形成长期稳定的相互作用,这表明这些化合物可能对 PIM-2 具有强大的抑制作用。本研究为 PIM-2 抑制剂研究提供了一种高效的生成模型,并发现了 5 种潜在的新型 PIM-2 抑制剂。





























 京公网安备 11010802027423号
京公网安备 11010802027423号