当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Intermolecular interaction energies with AROFRAG–A systematic approach for fragmentation of aromatic molecules
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-07-01 , DOI: 10.1002/jcc.27429 Emran Masoumifeshani 1 , Tatiana Korona 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-07-01 , DOI: 10.1002/jcc.27429 Emran Masoumifeshani 1 , Tatiana Korona 1
Affiliation
|
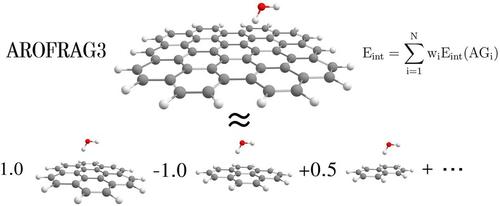
Intermolecular interactions with polycyclic aromatic hydrocarbons (PAHs) represent an important area of physisorption studies. These investigations are often hampered by a size of interacting PAHs, which makes the calculation prohibitively expensive. Therefore, methods designed to deal with large molecules could be helpful to reduce the computational costs of such studies. Recently we have introduced a new systematic approach for the molecular fragmentation of PAHs, denoted as AROFRAG, which decomposes a large PAH molecule into a set of predefined small PAHs with a benzene ring being the smallest unbreakable unit, and which in conjunction with the Molecules-in-Molecules (MIM) approach provides an accurate description of total molecular energies. In this contribution we propose an extension of the AROFRAG, which provides a description of intermolecular interactions for complexes composed of PAH molecules. The examination of interaction energy partitioning for various test cases shows that the AROFRAG3 model connected with the MIM approach accurately reproduces all important components of the interaction energy. An additional important finding in our study is that the computationally expensive long-range electron-correlation part of the interaction energy, that is, the dispersion component, is well described at lower AROFRAG levels even without MIM, which makes the latter models interesting alternatives to existing methods for an accurate description of the electron-correlated part of the interaction energy.
中文翻译:

AROFRAG 的分子间相互作用能——芳香族分子裂解的系统方法
多环芳烃 (PAH) 的分子间相互作用是物理吸附研究的一个重要领域。这些研究常常受到相互作用的多环芳烃的大小的阻碍,这使得计算成本极其昂贵。因此,设计用于处理大分子的方法可能有助于降低此类研究的计算成本。最近,我们引入了一种新的多环芳烃分子裂解系统方法,称为 AROFRAG,它将大的多环芳烃分子分解成一组预定义的小多环芳烃,其中苯环是最小的不易破碎的单元,并且与分子结合 -分子内 (MIM) 方法提供了对分子总能量的准确描述。在这篇文章中,我们提出了 AROFRAG 的扩展,它提供了由 PAH 分子组成的复合物的分子间相互作用的描述。对各种测试用例的相互作用能量分配的检查表明,与 MIM 方法相连接的 AROFRAG3 模型准确地再现了相互作用能量的所有重要组成部分。我们研究中的另一个重要发现是,即使没有 MIM,计算量大的相互作用能的远程电子关联部分(即色散分量)也可以在较低的 AROFRAG 水平下得到很好的描述,这使得后一种模型成为有趣的替代模型现有方法可以准确描述相互作用能的电子相关部分。
更新日期:2024-07-01
中文翻译:
AROFRAG 的分子间相互作用能——芳香族分子裂解的系统方法
多环芳烃 (PAH) 的分子间相互作用是物理吸附研究的一个重要领域。这些研究常常受到相互作用的多环芳烃的大小的阻碍,这使得计算成本极其昂贵。因此,设计用于处理大分子的方法可能有助于降低此类研究的计算成本。最近,我们引入了一种新的多环芳烃分子裂解系统方法,称为 AROFRAG,它将大的多环芳烃分子分解成一组预定义的小多环芳烃,其中苯环是最小的不易破碎的单元,并且与分子结合 -分子内 (MIM) 方法提供了对分子总能量的准确描述。在这篇文章中,我们提出了 AROFRAG 的扩展,它提供了由 PAH 分子组成的复合物的分子间相互作用的描述。对各种测试用例的相互作用能量分配的检查表明,与 MIM 方法相连接的 AROFRAG3 模型准确地再现了相互作用能量的所有重要组成部分。我们研究中的另一个重要发现是,即使没有 MIM,计算量大的相互作用能的远程电子关联部分(即色散分量)也可以在较低的 AROFRAG 水平下得到很好的描述,这使得后一种模型成为有趣的替代模型现有方法可以准确描述相互作用能的电子相关部分。






























 京公网安备 11010802027423号
京公网安备 11010802027423号