当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Machine Learning Force Field beyond the Limits of Classical and First-Principles Molecular Dynamics Simulations: The Case of Kaolinite Hydration
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-06-26 , DOI: 10.1021/acs.jpcc.4c03288 David Dell’Angelo 1 , Juliette Lainé 2 , Halima Said 1 , Yann Foucaud 3 , Michael Badawi 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-06-26 , DOI: 10.1021/acs.jpcc.4c03288 David Dell’Angelo 1 , Juliette Lainé 2 , Halima Said 1 , Yann Foucaud 3 , Michael Badawi 1
Affiliation
|
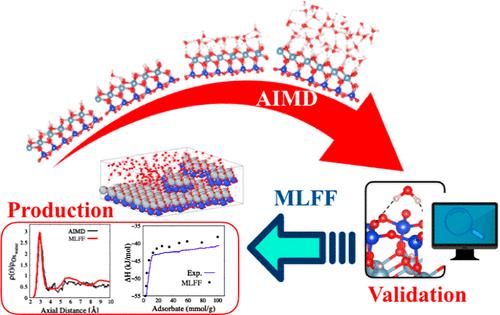
The understanding of the interaction between mineral surfaces and water holds great significance in unraveling the physical chemistry of surfaces in beneficiation processes. Within this study, we employ theoretical simulations to explore the adsorption mechanism of water molecules on the kaolinite surface, a clay mineral commonly found in iron ores. Albeit ab initio simulations offer key insights into the microscopic aspects of water adsorption, such as dissociation mechanisms or the hydrophilic/hydrophobic nature of the surface, the analysis of water structuring is limited by insufficient statistical sampling beyond the interfacial monolayer. By extending size and simulation time, the machine learning (ML) potential model is able to reproduce the experimental enthalpy profile by increasing the hydration film and provides converged densities with a well-defined minimum between the first water layer and water molecules beyond. Furthermore, the time correlation function combined with the ML method allows for the observation of dynamic occurrences on surfaces, including residence lifetime and the exchange of water molecules between hydration layers. In addition to offering valuable insights into the hydration mechanisms of kaolinite surfaces, for the first time we validate the use of the ML force field to overcome the limitations of both first-principles and classical molecular dynamics simulations for investigating adsorption phenomena.
中文翻译:

超越经典和第一原理分子动力学模拟极限的机器学习力场:高岭石水合案例
了解矿物表面与水之间的相互作用对于揭示选矿过程中表面的物理化学具有重要意义。在这项研究中,我们采用理论模拟来探索水分子在高岭石表面的吸附机制,高岭石是铁矿石中常见的粘土矿物。尽管从头开始模拟提供了对水吸附微观方面的关键见解,例如解离机制或表面的亲水/疏水性质,但水结构的分析由于界面单层之外的统计采样不足而受到限制。通过扩展尺寸和模拟时间,机器学习 (ML) 势模型能够通过增加水化膜来重现实验焓分布,并提供第一水层和后面的水分子之间具有明确定义的最小值的收敛密度。此外,时间相关函数与 ML 方法相结合,可以观察表面的动态变化,包括停留寿命和水化层之间水分子的交换。除了对高岭石表面的水化机制提供有价值的见解外,我们还首次验证了 ML 力场的使用,以克服第一原理和经典分子动力学模拟在研究吸附现象时的局限性。
更新日期:2024-06-26
中文翻译:
超越经典和第一原理分子动力学模拟极限的机器学习力场:高岭石水合案例
了解矿物表面与水之间的相互作用对于揭示选矿过程中表面的物理化学具有重要意义。在这项研究中,我们采用理论模拟来探索水分子在高岭石表面的吸附机制,高岭石是铁矿石中常见的粘土矿物。尽管从头开始模拟提供了对水吸附微观方面的关键见解,例如解离机制或表面的亲水/疏水性质,但水结构的分析由于界面单层之外的统计采样不足而受到限制。通过扩展尺寸和模拟时间,机器学习 (ML) 势模型能够通过增加水化膜来重现实验焓分布,并提供第一水层和后面的水分子之间具有明确定义的最小值的收敛密度。此外,时间相关函数与 ML 方法相结合,可以观察表面的动态变化,包括停留寿命和水化层之间水分子的交换。除了对高岭石表面的水化机制提供有价值的见解外,我们还首次验证了 ML 力场的使用,以克服第一原理和经典分子动力学模拟在研究吸附现象时的局限性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号