当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Monovalent cation binding to model systems and the macrocyclic depsipeptide, emodepside
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-06-26 , DOI: 10.1002/jcc.27451 Govindan Subramanian 1 , Kanika Manchanda 2 , Yirong Mo 3 , Rohit Y Sathe 4 , Prasad V Bharatam 5
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-06-26 , DOI: 10.1002/jcc.27451 Govindan Subramanian 1 , Kanika Manchanda 2 , Yirong Mo 3 , Rohit Y Sathe 4 , Prasad V Bharatam 5
Affiliation
|
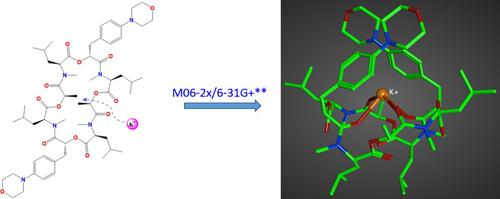
This study focuses on the systematic exploration of the emodepside conformations bound to monovalent K+ ion using quantum mechanical density functional theory (DFT) calculations at the M06-2X/6-31+G(d,p) level of theory. Nine conformers of emodepside and their complexes with K+ ion were characterized as stationary points on the potential energy surface. The conformational isomers were examined for their 3D structures, bonding, energetics, and interactions with the cation. A cavitand-like structure (CC) is identified to be the energetically most stable arrangement. To arrive at a better understanding of the K+ ion binding, calculations were initially performed on complexes formed by the K+ and Na+ ions with model ligands (methyl ester and N,N-dimethyl acetamide). Both the natural bond orbital (NBO) method and the block-localized wavefunction (BLW) energy decomposition approach was employed to assess the bonding and energetic contributions stabilizing the ion-bound model complexes. Finally, the solvent effect was evaluated through complete geometry optimizations and energy minimizations for the model ion-ligand complexes and the emodepside-K+ bound complexes using an implicit solvent model mimicking water and DMSO.
中文翻译:

单价阳离子与模型系统和大环缩酚肽、艾默德赛的结合
本研究重点是在 M06-2X/6-31+G(d,p) 理论水平上使用量子力学密度泛函理论 (DFT) 计算对与单价 K +离子结合的 emodepside 构象进行系统探索。艾莫德苷及其与 K +离子的复合物的九个构象异构体被表征为势能表面上的驻点。检查了构象异构体的 3D 结构、键合、能量以及与阳离子的相互作用。类空配体结构( CC )被认为是能量上最稳定的排列。为了更好地理解 K +离子结合,首先对 K +和 Na +离子与模型配体(甲酯和 N,N-二甲基乙酰胺)形成的复合物进行计算。采用自然键轨道(NBO)方法和块局域波函数(BLW)能量分解方法来评估稳定离子结合模型复合物的键合和能量贡献。最后,使用模拟水和 DMSO 的隐式溶剂模型,通过模型离子-配体复合物和 emodepside-K +结合复合物的完整几何优化和能量最小化来评估溶剂效应。
更新日期:2024-06-26
中文翻译:
单价阳离子与模型系统和大环缩酚肽、艾默德赛的结合
本研究重点是在 M06-2X/6-31+G(d,p) 理论水平上使用量子力学密度泛函理论 (DFT) 计算对与单价 K +离子结合的 emodepside 构象进行系统探索。艾莫德苷及其与 K +离子的复合物的九个构象异构体被表征为势能表面上的驻点。检查了构象异构体的 3D 结构、键合、能量以及与阳离子的相互作用。类空配体结构( CC )被认为是能量上最稳定的排列。为了更好地理解 K +离子结合,首先对 K +和 Na +离子与模型配体(甲酯和 N,N-二甲基乙酰胺)形成的复合物进行计算。采用自然键轨道(NBO)方法和块局域波函数(BLW)能量分解方法来评估稳定离子结合模型复合物的键合和能量贡献。最后,使用模拟水和 DMSO 的隐式溶剂模型,通过模型离子-配体复合物和 emodepside-K +结合复合物的完整几何优化和能量最小化来评估溶剂效应。






























 京公网安备 11010802027423号
京公网安备 11010802027423号