当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Sensitivity Analysis of Electrochemical Double Layer Approximations on Electrokinetic Predictions: Case Study for CO Reduction on Copper
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-06-25 , DOI: 10.1021/acs.jpcc.4c01457 Andrew Jark-Wah Wong 1 , Bolton Tran 2 , Naveen Agrawal 1 , Bryan R. Goldsmith 2 , Michael J. Janik 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-06-25 , DOI: 10.1021/acs.jpcc.4c01457 Andrew Jark-Wah Wong 1 , Bolton Tran 2 , Naveen Agrawal 1 , Bryan R. Goldsmith 2 , Michael J. Janik 1
Affiliation
|
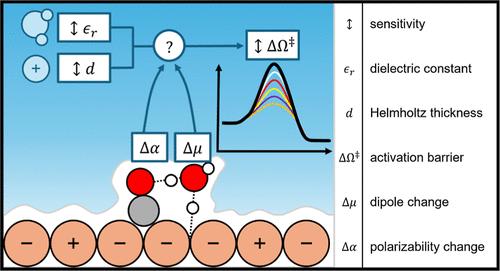
Density functional theory (DFT) modeling has been useful to electrocatalyst research, yet simulating the complexities of the electrode–electrolyte interface hinders progress in understanding reaction mechanisms and the underlying kinetics. Though many approaches to incorporating electrochemical double layer (EDL) features in DFT calculations have been developed, uncertainty in interfacial solvent properties and the distribution of ions leave the impact of the EDL on electrocatalytic kinetics unclear. Elucidating the sensitivity of DFT predictions to the EDL properties and model is crucial. Herein, we use an analytical Grand Canonical DFT framework (aGC-DFT) to quantify the sensitivity of potential-dependent activation energies to parameters of the EDL, incorporating a Helmholtz EDL model with varying dielectric constant (εr) and EDL width (d). We compute the activation barriers for OC–H, CO–H, and OC–CO bond formation from CO* on Cu. These elementary reactions are critical within the heavily debated reaction mechanism of CO2 reduction and are likely to impact overall activity and product selectivity. We show the aGC-DFT method produces consistent results with explicit GC-DFT calculations, while enabling probing of the EDL model sensitivity at a much lower computational cost. Reaction steps with significant dipole moment changes (i.e., CO–H bond formation) are highly sensitive to the chosen EDL parameters, such that the relative barriers of the OC–H, CO–H, and OC–CO bond formation steps depend significantly on EDL properties. Without knowledge of the interfacial properties of the EDL, there is substantial uncertainty in activation barriers and elementary reaction rates within a DFT analysis of electrocatalytic kinetics.
中文翻译:

电化学双层近似对电动预测的敏感性分析:铜上 CO 还原的案例研究
密度泛函理论(DFT)建模对于电催化剂研究很有用,但模拟电极-电解质界面的复杂性阻碍了理解反应机制和潜在动力学的进展。尽管已经开发了许多将电化学双层 (EDL) 特征纳入 DFT 计算的方法,但界面溶剂性质和离子分布的不确定性使得 EDL 对电催化动力学的影响尚不清楚。阐明 DFT 预测对 EDL 属性和模型的敏感性至关重要。在此,我们使用分析性大正则 DFT 框架 (aGC-DFT) 来量化电势相关激活能对 EDL 参数的敏感性,并结合具有不同介电常数 (ε r ) 的亥姆霍兹 EDL 模型和 EDL 宽度 (d)。我们计算了 Cu 上 CO* 形成 OC-H、CO-H 和 OC-CO 键的活化势垒。这些基元反应在备受争议的 CO 2 还原反应机制中至关重要,并且可能会影响整体活性和产物选择性。我们展示了 aGC-DFT 方法与显式 GC-DFT 计算产生一致的结果,同时能够以低得多的计算成本探测 EDL 模型灵敏度。具有显着偶极矩变化(即 CO-H 键形成)的反应步骤对所选的 EDL 参数高度敏感,因此 OC-H、CO-H 和 OC-CO 键形成步骤的相对势垒显着取决于EDL 属性。如果不了解 EDL 的界面特性,电催化动力学 DFT 分析中的活化势垒和基元反应速率存在很大的不确定性。
更新日期:2024-06-25
中文翻译:
电化学双层近似对电动预测的敏感性分析:铜上 CO 还原的案例研究
密度泛函理论(DFT)建模对于电催化剂研究很有用,但模拟电极-电解质界面的复杂性阻碍了理解反应机制和潜在动力学的进展。尽管已经开发了许多将电化学双层 (EDL) 特征纳入 DFT 计算的方法,但界面溶剂性质和离子分布的不确定性使得 EDL 对电催化动力学的影响尚不清楚。阐明 DFT 预测对 EDL 属性和模型的敏感性至关重要。在此,我们使用分析性大正则 DFT 框架 (aGC-DFT) 来量化电势相关激活能对 EDL 参数的敏感性,并结合具有不同介电常数 (ε r ) 的亥姆霍兹 EDL 模型和 EDL 宽度 (d)。我们计算了 Cu 上 CO* 形成 OC-H、CO-H 和 OC-CO 键的活化势垒。这些基元反应在备受争议的 CO 2 还原反应机制中至关重要,并且可能会影响整体活性和产物选择性。我们展示了 aGC-DFT 方法与显式 GC-DFT 计算产生一致的结果,同时能够以低得多的计算成本探测 EDL 模型灵敏度。具有显着偶极矩变化(即 CO-H 键形成)的反应步骤对所选的 EDL 参数高度敏感,因此 OC-H、CO-H 和 OC-CO 键形成步骤的相对势垒显着取决于EDL 属性。如果不了解 EDL 的界面特性,电催化动力学 DFT 分析中的活化势垒和基元反应速率存在很大的不确定性。





































 京公网安备 11010802027423号
京公网安备 11010802027423号