Nature Metabolism ( IF 18.9 ) Pub Date : 2024-06-25 , DOI: 10.1038/s42255-024-01076-x Martin Giera 1, 2 , Aries Aisporna 3 , Winnie Uritboonthai 3 , Gary Siuzdak 3, 4
|
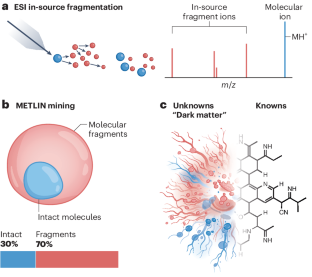
Mass spectrometry, specifically liquid chromatography–tandem mass spectrometry (LC–MS/MS), stands at the forefront of metabolomic, pharmaceutical and clinical analyses, offering high sensitivity and specificity in detecting metabolites and other small molecules within biological samples. Yet, this powerful technique has unveiled a puzzling abundance of unidentified spectral features, coined the ‘dark metabolome’1,2, which starkly contrasts with the known metabolic diversity. While humans are known to host approximately 20,000 protein encoding genes3,4, with only a subset of these expressing enzymes, the presumed chemical diversity indicated by LC–MS/MS suggests hundreds of thousands to millions of metabolites remain to be characterized2,5. This discrepancy raises a crucial point: might there be a technological explanation inflating the perceived complexity instead of a biological one?
The central dogma of biology describes the relation and information flow between the different omic layers from genome to transcriptome to proteome to metabolome, with the genome being the master code imprinted in any living system at its conception6. The code linking the first three layers is rather straightforward, with three nucleotides encoding one amino acid. However, such an encryption with the metabolome is yet to be revealed. This fact, combined with vast arrays of unannotated LC–MS/MS data, are two of the main reasons that the metabolome (being the entirety of small molecules within a biological system) cannot yet be defined. Current estimates suggest that less than 2% of observed LC–MS/MS spectra can be annotated, pointing to a potentially broad spectrum of unknown compounds2.
中文翻译:
代谢和化学质谱数据解释中源内碎片的隐藏影响
质谱分析,特别是液相色谱-串联质谱 (LC-MS/MS),处于代谢组学、药物和临床分析的前沿,在检测生物样品中的代谢物和其他小分子方面具有高灵敏度和特异性。然而,这项强大的技术揭示了大量令人困惑的未识别光谱特征,创造了“暗代谢组” 1,2 ,这与已知的代谢多样性形成鲜明对比。虽然已知人类拥有大约 20,000 个蛋白质编码基因3,4 ,但仅表达这些酶的一个子集,但 LC-MS/MS 表明的假定化学多样性表明,仍有数十万至数百万代谢物有待表征2,5 。这种差异提出了一个关键点:是否存在一种技术解释而不是生物学解释来夸大感知的复杂性?
生物学的中心法则描述了从基因组到转录组到蛋白质组再到代谢组的不同组学层之间的关系和信息流,基因组是在其概念中印刻在任何生命系统中的主代码6 。连接前三层的代码相当简单,三个核苷酸编码一个氨基酸。然而,这种代谢组加密尚未被揭示。这一事实与大量未注释的 LC-MS/MS 数据相结合,是代谢组(生物系统内所有小分子)尚无法定义的两个主要原因。目前的估计表明,只有不到 2% 的观察到的 LC-MS/MS 谱图可以进行注释,这表明未知化合物的谱谱可能很广2 。















































 京公网安备 11010802027423号
京公网安备 11010802027423号