当前位置:
X-MOL 学术
›
Ann. Neurol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Amyloid Neuropathy: From Pathophysiology to Treatment in Light-Chain Amyloidosis and Hereditary Transthyretin Amyloidosis
Annals of Neurology ( IF 8.1 ) Pub Date : 2024-06-24 , DOI: 10.1002/ana.26965
Pitcha Chompoopong 1 , Michelle L Mauermann 2 , Hasan Siddiqi 3 , Amanda Peltier 3, 4
Annals of Neurology ( IF 8.1 ) Pub Date : 2024-06-24 , DOI: 10.1002/ana.26965
Pitcha Chompoopong 1 , Michelle L Mauermann 2 , Hasan Siddiqi 3 , Amanda Peltier 3, 4
Affiliation
|
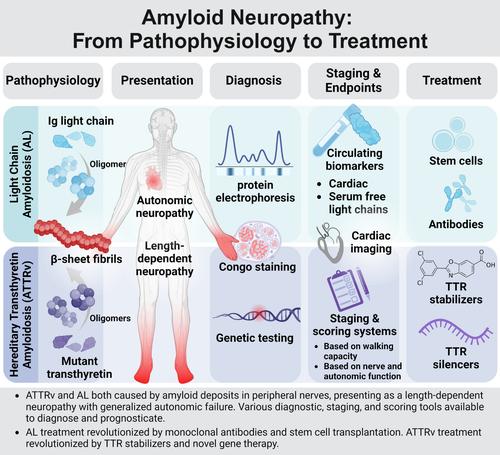
Amyloid neuropathy is caused by deposition of insoluble β-pleated amyloid sheets in the peripheral nervous system. It is most common in: (1) light-chain amyloidosis, a clonal non-proliferative plasma cell disorder in which fragments of immunoglobulin, light or heavy chain, deposit in tissues, and (2) hereditary transthyretin (ATTRv) amyloidosis, a disorder caused by autosomal dominant mutations in the TTR gene resulting in mutated protein that has a higher tendency to misfold. Amyloid fibrils deposit in the endoneurium of peripheral nerves, often extensive in the dorsal root ganglia and sympathetic ganglia, leading to atrophy of Schwann cells in proximity to amyloid fibrils and blood–nerve barrier disruption. Clinically, amyloid neuropathy is manifested as a length-dependent sensory predominant neuropathy associated with generalized autonomic failure. Small unmyelinated nerves are involved early and prominently in early-onset Val30Met ATTRv, whereas other ATTRv and light-chain amyloidosis often present with large- and small-fiber involvement. Nerve conduction studies, quantitative sudomotor axon testing, and intraepidermal nerve fiber density are useful tools to evaluate denervation. Amyloid deposition can be demonstrated by tissue biopsy of the affected organ or surrogate site, as well as bone-avid radiotracer cardiac imaging. Treatment of light-chain amyloidosis has been revolutionized by monoclonal antibodies and stem cell transplantation with improved 5-year survival up to 77%. Novel gene therapy and transthyretin stabilizers have revolutionized treatment of ATTRv, improving the course of neuropathy (less change in the modified Neuropathy Impairment Score + 7 from baseline) and quality of life. With great progress in amyloidosis therapies, early diagnosis and presymptomatic testing for ATTRv family members has become paramount. ANN NEUROL 2024;96:423–440
中文翻译:

淀粉样神经病:从病理生理学到轻链淀粉样变性和遗传性运甲状腺素蛋白淀粉样变性的治疗
淀粉样神经病是由不溶性β-折叠淀粉样蛋白片在周围神经系统中沉积引起的。最常见于:(1) 轻链淀粉样变性,一种克隆性非增殖性浆细胞疾病,其中免疫球蛋白轻链或重链片段沉积在组织中,以及 (2) 遗传性转甲状腺素蛋白 (ATTRv) 淀粉样变性,一种疾病由TTR基因的常染色体显性突变引起,导致突变蛋白具有更高的错误折叠倾向。淀粉样原纤维沉积在周围神经的神经内膜中,通常广泛分布于背根神经节和交感神经节,导致淀粉样原纤维附近的雪旺细胞萎缩和血神经屏障破坏。临床上,淀粉样神经病表现为与全身自主神经衰竭相关的长度依赖性感觉主导神经病。小的无髓鞘神经在早发性 Val30Met ATTRv 中早期且显着地受累,而其他 ATTRv 和轻链淀粉样变性通常表现为大纤维和小纤维受累。神经传导研究、定量催汗轴突测试和表皮内神经纤维密度是评估去神经的有用工具。淀粉样蛋白沉积可以通过受影响器官或替代部位的组织活检以及骨活性放射性示踪剂心脏成像来证明。单克隆抗体和干细胞移植彻底改变了轻链淀粉样变性的治疗,将 5 年生存率提高至 77%。新型基因疗法和运甲状腺素蛋白稳定剂彻底改变了 ATTRv 的治疗,改善了神经病变的病程(修正后的神经病变损伤评分 + 7 与基线相比变化较小)和生活质量。 随着淀粉样变性治疗的巨大进步,对 ATTRv 家族成员的早期诊断和症状前检测变得至关重要。安神经学 2024 年;96:423–440
更新日期:2024-06-24
中文翻译:
淀粉样神经病:从病理生理学到轻链淀粉样变性和遗传性运甲状腺素蛋白淀粉样变性的治疗
淀粉样神经病是由不溶性β-折叠淀粉样蛋白片在周围神经系统中沉积引起的。最常见于:(1) 轻链淀粉样变性,一种克隆性非增殖性浆细胞疾病,其中免疫球蛋白轻链或重链片段沉积在组织中,以及 (2) 遗传性转甲状腺素蛋白 (ATTRv) 淀粉样变性,一种疾病由TTR基因的常染色体显性突变引起,导致突变蛋白具有更高的错误折叠倾向。淀粉样原纤维沉积在周围神经的神经内膜中,通常广泛分布于背根神经节和交感神经节,导致淀粉样原纤维附近的雪旺细胞萎缩和血神经屏障破坏。临床上,淀粉样神经病表现为与全身自主神经衰竭相关的长度依赖性感觉主导神经病。小的无髓鞘神经在早发性 Val30Met ATTRv 中早期且显着地受累,而其他 ATTRv 和轻链淀粉样变性通常表现为大纤维和小纤维受累。神经传导研究、定量催汗轴突测试和表皮内神经纤维密度是评估去神经的有用工具。淀粉样蛋白沉积可以通过受影响器官或替代部位的组织活检以及骨活性放射性示踪剂心脏成像来证明。单克隆抗体和干细胞移植彻底改变了轻链淀粉样变性的治疗,将 5 年生存率提高至 77%。新型基因疗法和运甲状腺素蛋白稳定剂彻底改变了 ATTRv 的治疗,改善了神经病变的病程(修正后的神经病变损伤评分 + 7 与基线相比变化较小)和生活质量。 随着淀粉样变性治疗的巨大进步,对 ATTRv 家族成员的早期诊断和症状前检测变得至关重要。安神经学 2024 年;96:423–440
































 京公网安备 11010802027423号
京公网安备 11010802027423号