当前位置:
X-MOL 学术
›
Eur. J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Design, synthesis and antitumor activity of a novel FGFR2-selective degrader to overcome resistance of the FGFR2V564F gatekeeper mutation based on a pan-FGFR inhibitor
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2024-06-18 , DOI: 10.1016/j.ejmech.2024.116612
Zuli Hu 1 , Qiangsheng Zhang 1 , Zulong Li 1 , Hongling Yang 1 , Xin Chen 2 , Qi Zhang 1 , Tianqiong Yang 1 , Xiaojie He 1 , Qiang Feng 3 , Jun He 4 , Luoting Yu 1
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2024-06-18 , DOI: 10.1016/j.ejmech.2024.116612
Zuli Hu 1 , Qiangsheng Zhang 1 , Zulong Li 1 , Hongling Yang 1 , Xin Chen 2 , Qi Zhang 1 , Tianqiong Yang 1 , Xiaojie He 1 , Qiang Feng 3 , Jun He 4 , Luoting Yu 1
Affiliation
|
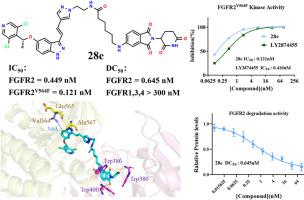
Aberrant activation of fibroblast growth factor receptors (FGFRs) contributes to the development and progression of multiple types of cancer. Although many FGFR inhibitors have been approved by the FDA, their long-term therapeutic efficacy is hampered by acquired resistance to gatekeeper mutations and low subtype selectivity. FGFR2 has been found to be frequently amplified or mutated in many tumors. In this study, we designed several PROTACs with different E3 ligands based on LY2874455. By screening the length of the linker and the binding site in various degraders, we obtained a novel and highly efficient FGFR2-selective degrader (DC = 0.645 nM, DC = 86 %). Compound selectively degraded FGFR2 and essentially avoided degradation of FGFR1,3,4 isoforms (DC > 300 nM). Compound significantly inhibited the proliferation of FGFR2-overexpressing cell lines, including KATOIII, SNU16, and AN3CA (IC = 0.794 nM/0.207 nM/4.626 nM), comparable to parental inhibitors. At the same time, the preferred compound showed superiority over the parental inhibitor in kinase inhibitory activity against the gatekeeper mutant isoform FGFR2 (IC = 0.121 nM). In summary, we identified as a novel selective degrader of FGFR2 with high potency and high potential to overcome resistance to gatekeeper mutation. The discovery of provides new evidence for the strategy of pan-inhibitor-based development of selective degrading agents.
中文翻译:

一种新型 FGFR2 选择性降解剂的设计、合成和抗肿瘤活性,以克服基于泛 FGFR 抑制剂的 FGFR2V564F 看门人突变的耐药性
成纤维细胞生长因子受体(FGFR)的异常激活导致多种癌症的发生和进展。尽管许多 FGFR 抑制剂已获得 FDA 批准,但其长期治疗效果因对看门突变的获得性耐药和低亚型选择性而受到阻碍。已发现 FGFR2 在许多肿瘤中频繁扩增或突变。在本研究中,我们基于LY2874455设计了几种具有不同E3配体的PROTAC。通过筛选各种降解剂中接头的长度和结合位点,我们获得了一种新型高效的 FGFR2 选择性降解剂(DC = 0.645 nM,DC = 86 %)。化合物选择性降解 FGFR2,并基本上避免 FGFR1,3,4 亚型的降解 (DC > 300 nM)。与亲本抑制剂相比,化合物显着抑制 FGFR2 过表达细胞系的增殖,包括 KATOIII、SNU16 和 AN3CA (IC = 0.794 nM/0.207 nM/4.626 nM)。同时,优选化合物对守门人突变同种型 FGFR2 的激酶抑制活性优于亲本抑制剂 (IC = 0.121 nM)。总之,我们确定了一种新型 FGFR2 选择性降解剂,具有高效能和高潜力来克服对守门突变的抗性。这一发现为基于泛抑制剂的选择性降解剂的开发策略提供了新的证据。
更新日期:2024-06-18
中文翻译:
一种新型 FGFR2 选择性降解剂的设计、合成和抗肿瘤活性,以克服基于泛 FGFR 抑制剂的 FGFR2V564F 看门人突变的耐药性
成纤维细胞生长因子受体(FGFR)的异常激活导致多种癌症的发生和进展。尽管许多 FGFR 抑制剂已获得 FDA 批准,但其长期治疗效果因对看门突变的获得性耐药和低亚型选择性而受到阻碍。已发现 FGFR2 在许多肿瘤中频繁扩增或突变。在本研究中,我们基于LY2874455设计了几种具有不同E3配体的PROTAC。通过筛选各种降解剂中接头的长度和结合位点,我们获得了一种新型高效的 FGFR2 选择性降解剂(DC = 0.645 nM,DC = 86 %)。化合物选择性降解 FGFR2,并基本上避免 FGFR1,3,4 亚型的降解 (DC > 300 nM)。与亲本抑制剂相比,化合物显着抑制 FGFR2 过表达细胞系的增殖,包括 KATOIII、SNU16 和 AN3CA (IC = 0.794 nM/0.207 nM/4.626 nM)。同时,优选化合物对守门人突变同种型 FGFR2 的激酶抑制活性优于亲本抑制剂 (IC = 0.121 nM)。总之,我们确定了一种新型 FGFR2 选择性降解剂,具有高效能和高潜力来克服对守门突变的抗性。这一发现为基于泛抑制剂的选择性降解剂的开发策略提供了新的证据。
































 京公网安备 11010802027423号
京公网安备 11010802027423号