当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Comparing Adsorption of an Electron-Rich Triphenylene Derivative: Metallic vs Graphitic Surfaces
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-06-19 , DOI: 10.1021/acs.jpcc.4c02376 Joris de la Rie 1 , Qiankun Wang 1 , Mihaela Enache 1 , Milan Kivala 2 , Meike Stöhr 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-06-19 , DOI: 10.1021/acs.jpcc.4c02376 Joris de la Rie 1 , Qiankun Wang 1 , Mihaela Enache 1 , Milan Kivala 2 , Meike Stöhr 1
Affiliation
|
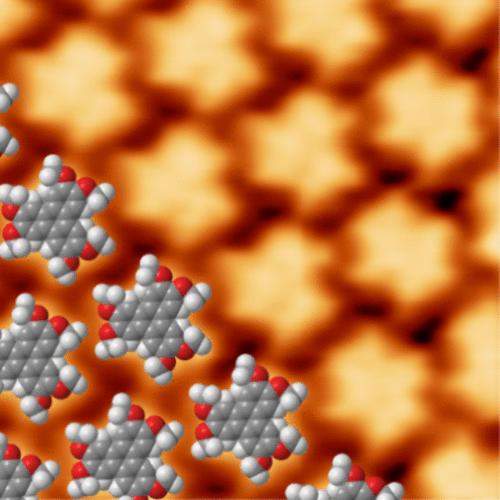
Crucial to the performance of devices based on organic molecules is an understanding of how the substrate–molecule interface influences both structural and electronic properties of the molecular layers. Within this context we studied the self-assembly of an alkoxy-triphenylene derived electron donor (HAT) in the monolayer regime on graphene/Ni(111). The molecules assembled into a close-packed hexagonal network commensurate with the graphene layer. Despite the commensurate structure, the HAT molecules only had a weak, physisorptive interaction with the substrate as pointed out by the photoelectron spectroscopy data. We discuss these findings in view of our recent reports for HAT adsorbed on Ag(111) and graphene/Ir(111). For all three substrates HAT adopts a similar close-packed hexagonal structure commensurate with the substrate while being physisorbed. The ionization potential is equal for all three substrates, supporting weak molecule–substrate interactions. These findings are remarkable, as commensurate overlayers usually only form at strongly interacting interfaces. We discuss potential reasons for this particular behavior of HAT which clearly sets itself apart from most studied molecule–substrate systems. In particular, these are the relatively weak but flexible intermolecular interactions, the molecular symmetry matching that of the substrate, and the comparatively weak but directional molecule–substrate interactions.
中文翻译:

比较富电子苯并菲衍生物的吸附:金属表面与石墨表面
对于基于有机分子的器件的性能至关重要的是了解基底-分子界面如何影响分子层的结构和电子特性。在此背景下,我们研究了烷氧基苯并菲衍生的电子给体(HAT)在石墨烯/Ni(111)单层体系中的自组装。这些分子组装成与石墨烯层相称的密堆积六边形网络。尽管具有相称的结构,但正如光电子能谱数据所指出的,HAT 分子与基底仅具有微弱的物理吸附相互作用。我们根据最近关于吸附在 Ag(111) 和石墨烯/Ir(111) 上的 HAT 的报告讨论了这些发现。对于所有三种基材,HAT 在物理吸附时均采用与基材相称的类似密堆积六方结构。所有三种底物的电离势相等,支持弱的分子-底物相互作用。这些发现是引人注目的,因为相应的覆盖层通常只在强相互作用的界面上形成。我们讨论了 HAT 这种特殊行为的潜在原因,它显然与大多数研究的分子-底物系统不同。特别是,这些是相对较弱但灵活的分子间相互作用,与底物匹配的分子对称性,以及相对较弱但有方向性的分子-底物相互作用。
更新日期:2024-06-21
中文翻译:
比较富电子苯并菲衍生物的吸附:金属表面与石墨表面
对于基于有机分子的器件的性能至关重要的是了解基底-分子界面如何影响分子层的结构和电子特性。在此背景下,我们研究了烷氧基苯并菲衍生的电子给体(HAT)在石墨烯/Ni(111)单层体系中的自组装。这些分子组装成与石墨烯层相称的密堆积六边形网络。尽管具有相称的结构,但正如光电子能谱数据所指出的,HAT 分子与基底仅具有微弱的物理吸附相互作用。我们根据最近关于吸附在 Ag(111) 和石墨烯/Ir(111) 上的 HAT 的报告讨论了这些发现。对于所有三种基材,HAT 在物理吸附时均采用与基材相称的类似密堆积六方结构。所有三种底物的电离势相等,支持弱的分子-底物相互作用。这些发现是引人注目的,因为相应的覆盖层通常只在强相互作用的界面上形成。我们讨论了 HAT 这种特殊行为的潜在原因,它显然与大多数研究的分子-底物系统不同。特别是,这些是相对较弱但灵活的分子间相互作用,与底物匹配的分子对称性,以及相对较弱但有方向性的分子-底物相互作用。





































 京公网安备 11010802027423号
京公网安备 11010802027423号